Clinical Description
The course of epidermolysis bullosa with pyloric atresia (EB-PA) is usually severe and often lethal in the neonatal period. Most affected children die as neonates due to mucosal erosions and blistering, pyloric stenosis or atresia, respiratory failure, or overwhelming infection.
Cutaneous manifestations. Those who survive the neonatal period may have severe blistering with formation of granulation tissue on the skin around the mouth, diaper area, nose, fingers, and toes, and internally around the trachea. However, some affected individuals have little or no blistering later in life.
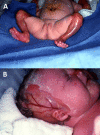
Congenital localized absence of skin (aplasia cutis congenita). Infants with extensive aplasia cutis congenita and blistering or erosions may have fatal infections with sepsis and severe electrolyte imbalance in the first weeks to months of life. Aplasia cutis congenita can be extensive in these neonates, especially on the extremities and sides of the scalp (see ). One review reported that 50% of individuals with EB-PA and aplasia cutis congenita died during the first months of life [
Martinez-Moreno et al 2020].
Mucosal erosions are predominantly oropharyngeal, but erosions have also been reported of the bladder, trachea, bowel, stomach, and esophagus [
Mylonas et al 2019].
Deformities of the ear and/or nose, such as ear hypoplasia
Milia
Nail dystrophy and granulation tissue of the nail, including changes in size, color, shape, or texture and subsequent loss of nails
Scarring alopecia. Complete loss of scalp hair follicles as a result of scarring
Hypotrichosis. Reduction in the number of hair follicles in a given area compared to the number of hair follicles in the same area of an unaffected individual of the same sex
Exuberant granulation tissue does not usually appear until early childhood, and most children with EB-PA do not survive beyond infancy.
Wound infections. Treatment of chronic wound infections is a challenge. Many affected individuals become infected with resistant bacteria, most often methicillin-resistant Staphylococcus aureus (MRSA), multidrug-resistant Pseudomonas aeruginosa, and group A beta-hemolytic Streptococci.
Child with epidermolysis bullosa with pyloric atresia and extensive aplasia cutis congenita of the extremities (A) and scalp (B). Reproduced with permission from Lucky et al [2021]
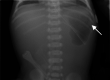
Pyloric atresia may be detected in utero as polyhydramnios via ultrasound or MRI (see Genetic Counseling, Prenatal Testing and Preimplantation Genetic Testing) or becomes evident at birth. It is characterized by vomiting, failure to tolerate any feeding or to pass stool, and a distended abdomen with a large stomach bubble (see ). Surgical repair of the pyloric atresia is necessary for survival. Surgical repair is most commonly performed with a Heineke-Mikulicz pyloroplasty (for type 1 pyloric atresia) or gastroduodenostomy (for type 2 pyloric atresia) [Mylonas et al 2019].
Renal and ureteral anomalies can include dysplastic/multicystic kidney, hydronephrosis/hydroureter, acute renal tubular necrosis, obstructive uropathy, ureterocele, duplicated renal collecting system, vesicoureteral reflux, interstitial nephritis, and/or absent bladder [Puvabanditsin et al 1997, Kambham et al 2000, Wallerstein et al 2000, Nakano et al 2001, Varki et al 2006, Pfendner et al 2007, Walker et al 2017, Mylonas et al 2019].
Protein-losing enteropathy has been reported in some individuals with EB-PA, resulting in large-volume diarrhea and poor weight gain [Verma et al 2020, Wee et al 2021, DeMaria et al 2022, Kaneyasu et al 2022]. Bowel biopsies show decreased integrin B4 [Wee et al 2021] as well as lamina lucida separation with hypoplastic hemidesmosomes [DeMaria et al 2022].
Ocular findings are seen in a minority of individuals but can include corneal blisters/erosions, conjunctivitis, and corneal opacification [Mylonas et al 2019].
Fluids/electrolytes/nutrition. Fluid and electrolyte issues can be significant and life threatening in the neonatal period. Infants with widespread disease require careful management.
In children who survive the newborn period, nutritional deficiencies can include the following:
Phenotype Correlations by Gene
ITGA6 pathogenic variants are associated with a poor prognosis [Masunaga et al 2017, Mylonas et al 2019].
ITGB4 pathogenic variants are associated with a more variable prognosis than either ITGA6- or PLEC-related EB-PA.
PLEC pathogenic variants are associated with a poor prognosis, and the majority of infants die as neonates or in the first year [Kaneyasu et al 2022, Pongmee et al 2022]. Individuals with PLEC pathogenic variants have been reported to have cardiomyopathy [Vahidnezhad et al 2022], but there are no reports of cardiomyopathy occurring in an individual with pyloric atresia.
Genotype-Phenotype Correlations
ITGB4. The most severe cutaneous manifestations are caused by biallelic pathogenic variants that result in a premature termination codon, although pathogenic variants between exon 3 and intron 11 are also associated with a poor prognosis. A more favorable prognosis is typically associated with missense variants and in-frame insertions or deletions. However, missense variants in the plectin-binding region or in cysteine-rich domains are associated with a poor prognosis. Several additional missense variants result in a severe phenotype, such as the recurrent ITGB4 variant p.Cys61Tyr, which is common in Hispanic individuals with EB-PA [Varki et al 2006, Masunaga et al 2015, Mutlu et al 2015, Mencía et al 2016, Mylonas et al 2019].
Nomenclature
Although ITGA6-, ITGB4-, and PLEC-related EB-PA are clinically quite similar, in the 2020 epidermolysis bullosa (EB) classification system [Has et al 2020], individuals with EB-PA due to PLEC pathogenic variants are considered to have EB simplex and those with EB-PA due to ITGB4 or ITGA6 pathogenic variants are considered to have junctional EB.
ITGA6- and ITGB4-related EB-PA may also be referred to as Carmi syndrome or junctional epidermolysis bullosa with pyloric atresia (JEB-PA).
Prevalence
EB-PA is rare, and its prevalence has not been determined. However, it can be conservatively estimated at fewer than one in 5,000 (~10 times rarer than severe junctional EB [previously called Herlitz junctional EB], the carrier frequency of which is ~1:700).