Clinical Description
Prior to the molecular characterization of Floating-Harbor syndrome (FHS) by Hood et al [2012], a number of reports included descriptions of individuals in whom the diagnosis of FHS could be questioned. This GeneReview only includes information on those 73 individuals with molecularly confirmed FHS (i.e., presence of a heterozygous SRCAP pathogenic variant) [Hood et al 2012, Le Goff et al 2013, Nikkel et al 2013, Dong et al 2014, Kehrer et al 2014, Nagasaki et a 2014, Seifert et al 2014, Amita et al 2016, Coughlin et al 2017, Singh et al 2017, Budisteanu et al 2018, Choi et al 2018, Milani et al 2018, Shields et al 2019]. The 41 females and 32 males range in age from eight months to 52 years.
FHS is frequently recognized in early childhood because of the characteristic facial features (). Infants and younger children are often referred for assessment of poor growth or developmental (predominantly speech and language) delay.
Craniofacial features include triangular face, deep-set eyes, short philtrum, wide mouth with a thin vermilion border of the upper lip, long nose with a narrow bridge, broad base, broad tip, and low-hanging columella, and low-set ears.
The features become more pronounced with age, especially the length of the nose and the width of the nasal tip.
Intellect. Although gross motor and fine motor milestones are within normal limits, affected individuals typically have mild-to-moderate intellectual disability. A disorder of speech and language is the most severe disability. Most aspects of communication are affected; expressive language is most consistently and severely affected. Dysarthria and verbal dyspraxia with phoneme imprecision is most common, with absent speech in some individuals. Voice is described as hypernasal and high-pitched. The majority of affected children receive mainstream education with individualized educational plans. Regression of skills is not typical of FHS.
Behavior. Many individuals with FHS have temperament and behavior differences and difficulties: temper tantrums in infancy and attention-deficit/hyperactivity disorder spectrum with impulsivity, inattention, and restlessness at school age. Aggressive and violent outbursts can occur. Obsessive compulsive disorder and anxiety have been observed. Behavior problems are reported to improve in adulthood.
Growth. Short stature is a cardinal sign of FHS. The majority of individuals with FHS have low birth weight (from 3 SD below the mean to 0 SD) and normal head circumference (2 SD below the mean to 0 SD). In the first years of life weight gain and linear growth are poor. A significant delay in bone age is reported (≥2 SD below the mean) with normalization between ages six and 12 years. Average adult height is 140-155 cm.
Puberty. Early puberty has been reported; data are insufficient to determine the incidence in either sex.
Eye. Five of 73 individuals have been reported with hyperopia and eight of 13 with strabismus. One individual had anterior chamber abnormalities.
Hearing. Conductive hearing loss has been seen in eleven of 73 individuals with FHS. Cochlear abnormality has been observed in one of 73.
Neurologic. Seizures have been observed in seven of 73 individuals.
Gastrointestinal. Reflux can be severe, requiring G-tube feeding in some. Constipation and colonic strictures have been observed. One of 73 individuals had celiac disease; two had transient gluten intolerance.
Genitourinary. Renal and genitourinary anomalies can occur and include hypospadias and undescended testes, epididymal cysts, varicocele, and posterior urethral valves in boys. Hydronephrosis/renal pelviectasis and nephrocalcinosis, renal cysts, and renal agenesis have been observed. One adult of the 73 reported individuals developed polycystic kidney disease and end-stage renal disease.
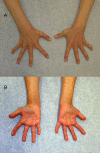
Orthopedic. The body habitus is often stocky with a broad chest and short neck. Additional features include hand anomalies such as clinodactyly, brachydactyly, short thumbs, and broad fingertips that give the appearance of clubbing (). Clavicular anomalies including pseudarthrosis and clavicular hypoplasia have been observed, as have short metacarpals, 11 pairs of ribs, kyphoscoliosis, prominent joints, dysplastic hips, and dislocated radial heads. Perthes disease has also been reported.
Dental. A number of individuals with FHS have dental problems (e.g., caries, microdontia, oligodontia, delayed loss of primary teeth) and orthodontic problems (e.g., maxillary retrusion, underbite).
Cardiac. Cardiac malformations are not usually a feature of FHS. Of 73 affected individuals one had mild aortic coarctation, one had mesocardia with persistent left superior vena cava, two had atrial septal defect, and one individual had tetralogy of Fallot.