Summary
Clinical characteristics.
Long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency and trifunctional protein (TFP) deficiency are caused by impairment of mitochondrial TFP. TFP has three enzymatic activities – long-chain enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase, and long-chain 3-ketoacyl-CoA thiolase. In individuals with LCHAD deficiency, there is isolated deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase, while deficiency of all three enzymes occurs in individuals with TFP deficiency.
Individuals with TFP deficiency can present with a severe-to-mild phenotype, while individuals with LCHAD deficiency typically present with a severe-to-intermediate phenotype.
- Neonates with the severe phenotype present within a few days of birth with hypoglycemia, hepatomegaly, encephalopathy, and often cardiomyopathy.
- The intermediate phenotype is characterized by hypoketotic hypoglycemia precipitated by infection or fasting in infancy.
- The mild (late-onset) phenotype is characterized by myopathy and/or neuropathy.
Long-term complications include peripheral neuropathy and retinopathy.
Diagnosis/testing.
The diagnosis of LCHAD/TFP deficiency is established in a proband with elevation of long-chain 3-hydroxyacylcarnitine species in plasma and/or increased excretion of 3-hydroxy-dicarboxylic acids in urine in combination with identification of biallelic pathogenic variants in HADHA or HADHB by molecular genetic testing.
Distinguishing LCHAD deficiency from TFP deficiency requires identification of isolated long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency on enzymatic assay in lymphocytes or skin fibroblasts. TFP deficiency is confirmed by the identification of deficiencies in all three TFP enzymatic activities (long-chain enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase, and long-chain 3-ketoacyl-CoA thiolase) in lymphocytes or skin fibroblasts.
Management.
Treatment: Avoidance of fasting using frequent feeds, decreasing feeding intervals and supplemental carbohydrates during illness, and continuing overnight feeds in older children as needed for hypoglycemia; medium-chain triglyceride (MCT) or triheptanoin supplementation; low-fat diet; carnitine supplementation in those with carnitine deficiency; feeding therapy and gastrostomy tube as needed; developmental services; and treatment of cardiac dysfunction, peripheral neuropathy, and retinopathy by relevant specialists. Emergency outpatient treatment for mild decompensation includes decreasing the fasting interval, administration of antipyretics for fever, and antiemetics as needed for vomiting. Acute treatment includes hospitalization with intravenous fluid containing at least 10% dextrose, and bicarbonate therapy for severe metabolic acidosis; management of hyperammonemia and rhabdomyolysis; and management of cardiomyopathy per cardiologist.
Prevention of primary manifestations: Avoidance of fasting; supplementation with MCT or triheptanoin; strict dietary management; education of parents and caregivers to ensure prompt treatment; written protocol for emergency treatment.
Surveillance: Monitor nutrition, serum plasma free and total carnitine, acylcarnitine profile, creatine kinase, AST, and ALT with frequency based on age; annual comprehensive fatty acid profile; monitor head size, growth, and development at each visit throughout childhood; neuropsychological testing and quality of life assessments as needed; EKG and echocardiography annually or more frequently as needed; annual neurology evaluation with nerve conduction velocity and electromyography as needed; annual ophthalmology evaluation with electroretinography every two to three years.
Agents/circumstances to avoid: Fasting; inadequate calories during stressors; dehydration; high-fat diets including ketogenic and carbohydrate restricted diet; anesthetics that contain high doses of long-chain fatty acids; intravenous intralipids during acute metabolic crisis.
Evaluation of relatives at risk: Testing of all at-risk sibs of any age is warranted (targeted molecular genetic testing if the familial pathogenic variants are known or plasma acylcarnitine profile, plasma free and total carnitine, and urine organic acid assay if the pathogenic variants in the family are not known) to allow for early diagnosis and treatment of LCHAD/TFP deficiency.
Pregnancy management: Increase MCT intake in the third trimester; high dextrose infusion in the peripartum period. Monitor for HELLP syndrome and acute fatty liver of pregnancy in pregnant females who are heterozygous for an HADHA or HADHB pathogenic variant (including suspected carriers).
Genetic counseling.
LCHAD/TFP deficiency is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an HADHA or HADHB pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial pathogenic variants. Once the HADHA or HADHB pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal and preimplantation genetic testing are possible.
GeneReview Scope
Table.
Synonyms and Included Genes
Diagnosis
No consensus clinical diagnostic criteria for long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency or trifunctional protein (TFP) deficiency have been published.
Suggestive Findings
Scenario 1: Abnormal Newborn Screening (NBS) Result
NBS for LCHAD/TFP deficiency is primarily based on quantification of the analytes 3-hydroxypalmitoyl carnitine (C16-OH) and 3-hydroxyoleoylcarnitine (C18:1-OH) on dried blood spots.
C16-OH and C18:1-OH values above the cutoff reported by the screening laboratory are considered positive and require follow-up biochemical testing including plasma acylcarnitine and urine organic acid profiles.
If the follow-up biochemical testing supports the likelihood of LCHAD/TFP deficiency, additional testing is required to establish the diagnosis (see Establishing the Diagnosis).
The following medical interventions need to begin immediately on receipt of an abnormal NBS result while additional testing is performed to determine whether this is a true positive NBS result and to establish a definitive diagnosis of LCHAD/TFP deficiency:
- Evaluation of the newborn to ascertain clinical status
- Education of the caregivers to avoid prolonged fasting and to monitor for decreased oral intake, vomiting, or lethargy
- Immediate intervention (to be considered if the newborn is not doing well clinically) possibly including admission to the hospital, fluid resuscitation, infusion of IV dextrose (10% or higher), and cardiac evaluation
Scenario 2: Symptomatic Individual
Supportive – but nonspecific – clinical findings, laboratory findings, and family history include the following.
Clinical findings
- Neonatal onset (severe)
- Hypoketotic hypoglycemia, hepatomegaly
- Cardiomyopathy
- Encephalopathy
- Infantile onset (intermediate). Recurrent hypoketotic hypoglycemia precipitated by infection or fasting
- Late onset (mild)
- Episodic rhabdomyolysis
- Exercise intolerance and muscle weakness
- Peripheral neuropathy
- Retinopathy
Supportive laboratory findings
- Nonspecific:
- Hypoglycemia (nonketotic or hypoketotic) with blood glucose often <45 mg/dL
- Urinalysis that demonstrates the absence of ketones in the setting of hypoglycemia
- Metabolic acidosis
- Lactic acidosis
- Hyperammonemia: blood ammonia level may be >200 µmol/L in newborns and >100 µmol/L after the neonatal period
- Elevated liver transaminases (AST, ALT)
- Elevated creatine kinase (CK), particularly in the late-onset myopathic form. A CK value greater than five times the upper limit of reference is suggestive of rhabdomyolysis (range 1,000-100,000 IU/L). A CK value of >15,000 IU/L at presentation increases the risk for acute kidney injury [Bosch et al 2009].
- Specific:
- Plasma acylcarnitine profile. The elevation of 3-hydroxy derivatives of C16, C18, and C18:1 is highly suggestive of LCHAD/TFP deficiency. The plasma acylcarnitine profile typically shows elevations of C16-OH, C18-OH, C18:1-OH, and elevated ratios of C16-OH/C16 and C18-OH/C18.
- Urine organic acid analysis. Elevations of 3-hydroxy-dicarboxylic acids and lactic acid
Note: Because elevations of these metabolites can be intermittent particularly in individuals with milder disease, follow-up testing is required to establish the diagnosis of LCHAD/TFP deficiency (see Establishing the Diagnosis) [Elizondo et al 2020].
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of LCHAD deficiency is established in a proband with elevation of long-chain 3-hydroxyacylcarnitine species in plasma and/or increased excretion of 3-hydroxy-dicarboxylic acids in urine in combination with identification of biallelic pathogenic (or likely pathogenic) variants in HADHA by molecular genetic testing (see Table 1). Distinguishing LCHAD deficiency from TFP deficiency requires identification of isolated long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency on enzymatic assay in lymphocytes or skin fibroblasts.
The diagnosis of TFP deficiency is established in a proband with elevation of long-chain 3-hydroxyacylcarnitine species in plasma and/or increased excretion of 3-hydroxy-dicarboxylic acids in urine in combination with identification of biallelic pathogenic (or likely pathogenic) variants in HADHA or HADHB by molecular genetic testing (see Table 1). Distinguishing TFP deficiency from LCHAD deficiency requires identification of deficiency in all three TFP enzymatic activities (long-chain enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase, and long-chain 3-ketoacyl-CoA thiolase) in lymphocytes or skin fibroblasts.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Most affected individuals have an abnormal acylcarnitine profile. An individual with persistent abnormal acylcarnitine profile is presumed to have LCHAD/TFP deficiency even if only one pathogenic variant is identified.
Molecular Genetic Testing Approaches
Scenario 1: Abnormal newborn screening (NBS) result. When NBS results and other laboratory findings suggest the diagnosis of LCHAD/TFP deficiency, molecular genetic testing approaches can include single-gene testing or use of a multigene panel.
- Serial single-gene testing. Sequence analysis detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
- Perform sequence analysis of HADHA first. If only one pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
- If HADHA testing is negative, perform sequence analysis of HADHB. If only one pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
- A multigene panel that includes HADHA, HADHB, and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Scenario 2: Symptomatic individual. For a symptomatic individual who has findings associated with late-onset TFP deficiency OR neonatal-onset LCHAD/TFP deficiency that has not been treated (because symptoms occurred before NBS results were returned, NBS was not performed, or NBS yielded a false negative result), molecular genetic testing approaches can include serial single-gene testing or use of a multigene panel.
When the diagnosis of LCHAD/TFP deficiency has not been considered, comprehensive genomic testing, which does not require the clinician to determine which gene(s) are likely involved, is an option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in LCHAD/TFP Deficiency
Biochemical Testing Approaches
In vitro probe analysis. Skin fibroblasts incubated with palmitic acid and culture medium can be assayed for acylcarnitine after 96 hours of incubation. In individuals with LCHAD/TFP deficiency there is substantial accumulation of C16-OH [Okun et al 2002].
Clinical Characteristics
Clinical Description
Long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency and trifunctional protein (TFP) deficiency are caused by impairment of mitochondrial TFP. TFP has three enzymatic activities – long-chain enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase, and long-chain 3-ketoacyl-CoA thiolase. Deficiency of the enzyme long-chain 3-hydroxyacyl-CoA dehydrogenase occurs in individuals with LCHAD deficiency, while deficiency of all three enzymes occurs in individuals with TFP deficiency.
LCHAD and TFP deficiency are disorders of long-chain fatty acid oxidation, which typically present with recurrent episodes of hypoketotic hypoglycemia precipitated by fasting or illness. In addition, the other characteristic manifestations of long-chain fatty acid oxidation defects (FAODs) such as cardiomyopathy, liver dysfunction, or rhabdomyolysis may be present. However, peripheral neuropathy and retinopathy are unique complications of these disorders not seen in other FAODs. The clinical presentation represents a continuous spectrum of severity ranging from severe neonatal-onset to mild late-onset forms. Individuals with LCHAD deficiency usually present with a severe-to-intermediate phenotype, while individuals with TFP deficiency typically present with a severe-to-mild phenotype.
Table 2.
LCHAD/TFP Deficiency: Frequency of Select Features
Neonatal Onset (Severe/Cardiac Phenotype)
The neonatal-onset (severe/cardiac) presentation is more common in individuals with TFP deficiency than in those with LCHAD deficiency. The main manifestations are the following:
- Metabolic decompensation. Newborns present within a few days of birth with a Reye-like syndrome presentation: encephalopathy, hypoketotic hypoglycemia, hepatomegaly with elevated transaminases and hepatosteatosis, and lactic acidosis. Hyperammonemia may also be present. The metabolic decompensation is rapidly progressive and requires immediate intervention. The acute metabolic decompensation is often associated with liver dysfunction manifesting as hepatomegaly, elevated liver enzymes, or liver failure.
- Neurologic manifestations. Severe neonatal presentation characterized by hypoglycemia and liver dysfunction is usually associated with encephalopathy manifesting as lethargy, poor feeding, seizures, apnea, or coma.
- Cardiac manifestations. The severe form is associated with progressive dilated cardiomyopathy manifesting as arrhythmias and cardiac failure. It is associated with very high mortality.
Infantile Onset (Intermediate/Hepatic Phenotype)
Individuals with the intermediate or moderate severity phenotype present later in infancy. This is the most common presentation in LCHAD deficiency and relatively uncommon in TFP deficiency.
The classic presentation is acute metabolic decompensation precipitated by fasting or infection.
- The metabolic decompensation is characterized by hypoketotic hypoglycemia often associated with lactic acidosis, elevated liver enzymes, and high creatine kinase (CK).
- Infants may present with vomiting, lethargy, poor feeding, and hepatomegaly.
- Associated baseline findings including muscle weakness, feeding difficulties, and hypotonia may be present.
- Other manifestations of this form (more common in previously untreated individuals): cardiomyopathy (dilated or hypertrophic), long QT intervals, liver cirrhosis, cholestasis, developmental delays, and failure to thrive. Cardiomyopathy may be present at baseline or dilated cardiomyopathy may first appear during metabolic crisis even in previously treated individuals.
- Prompt diagnosis and initiation of treatment is crucial for reversal of cardiomyopathy and favorable outcome. Newborn screening has enabled presymptomatic diagnosis and thus improved outcome.
Late Onset (Mild/Neuromyopathic Phenotype)
Individuals with the mild phenotype usually present after infancy with neuromuscular symptoms. The isolated neuromyopathic presentation is typical of mild TFP deficiency and rare in LCHAD deficiency. However, infants with LCHAD deficiency with the intermediate/hepatic phenotype may present later with neuromyopathic symptoms. The common manifestations are the following:
- Skeletal myopathy manifests as muscle weakness, exercise intolerance, and hypotonia. Episodic rhabdomyolysis precipitated by prolonged exercise, cold exposure, fasting, or infection is characteristic of this phenotype. These episodes are characterized by diffuse muscle pain, profound weakness, myoglobinuria, and elevations of serum CK (>5x the upper limit of normal), aldolase, aspartate aminotransferase, and alanine transaminase.
- Neuropathy. Many individuals present with progressive peripheral neuropathy resembling axonal Charcot-Marie-Tooth disease [Immonen et al 2016a, Grünert et al 2021].
Long-Term Complications
Long-term complications in those with the intermediate and late-onset phenotypes include the following:
- Peripheral neuropathy is a unique long-term complication of LCHAD/TFP deficiency. Age of onset ranges from infancy to adulthood (median: age ~7 years) [Grünert et al 2021]. Onset is earlier in individuals with TFP deficiency than in those with LCHAD deficiency. It is progressive and sensorimotor in nature. However, it can be pure sensory or pure motor. Polyneuropathy is described as axonal or axonal with secondary demyelination on electrophysiologic studies. Neuropathy can worsen during metabolic crisis. Early diagnosis and treatment can delay the onset but may not prevent this complication.
- Retinopathy, another unique complication, is much more common in individuals with LCHAD deficiency than those with TFP deficiency. It is progressive and correlates with disease severity. Four different stages of retinopathy in LCHAD deficiency have been described [Tyni et al 1998]:
- Stage 1. Normal to diffuse hypopigmentation of the fundus
- Stage 2. Pigment clumping in the fovea
- Stage 3. Macular pallor and migration of pigmentary changes toward the periphery
- Stage 4. Atrophy of the posterior fundus and further peripheral migration of pigmentary changes
Visual impairment is present from stage 3 onward. Hence, retinopathy may be missed if fundal imaging and electroretinogram are not done. Approximately half of individuals with LCHAD deficiency have evidence of retinopathy by age two years. Early diagnosis and treatment can slow the progress but may not prevent this complication [Fahnehjelm et al 2016].
Other
Rare manifestations in individuals with LCHAD/TFP deficiency include hypoparathyroidism, neonatal respiratory distress syndrome, and necrotizing enterocolitis [Tyni et al 1997, Diekman et al 2013, Karall et al 2015, van Vliet et al 2018].
Pregnancy Complications
Pregnancy complications such as HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome and acute fatty liver of pregnancy are seen in about 15%-25% of pregnancies in women carrying a fetus affected with LCHAD/TFP deficiency [den Boer et al 2002, Spiekerkoetter et al 2003, Karall et al 2015]. The pathophysiology of maternal complications is unclear. One hypothesis is that HELLP syndrome is precipitated by the excessive hydroxyacyl derivatives produced by the affected fetus [Kobayashi et al 2015]. An alternative hypothesis is that maternal heterozygosity for LCHAD/TFP deficiency causes hepatic insufficiency [Blish & Ibdah 2005].
Genotype-Phenotype Correlations
HADHA. Homozygous c.1528G>C variants are associated with LCHAD deficiency. Most individuals with LCHAD deficiency have at least one allele with this variant [Ijlst et al 1996]. In the largest cohort of individuals with LCHAD deficiency, c.1528G>C was present in 84 of 98 alleles. Only one individual was homozygous for another variant [den Boer et al 2002]. However, a few individuals who were compound heterozygous for this variant and another pathogenic variant in HADHA were reported to have TFP deficiency [Grünert et al 2021]. Enzymatic studies were not provided for those individuals. In the absence of homozygosity for this variant, enzyme assay is needed to distinguish between these conditions.
HADHB. In general, individuals with HADHB missense pathogenic variants present with milder phenotypes than those with premature termination or frameshift variants. A missense variant on at least one allele favors the milder phenotype. However, the amino acid p.Arg28 appears critical for TFP function/stability, and variants altering this amino acid lead to the severe presentation when in combination with a severe variant on the other allele [Spiekerkoetter et al 2003].
Although clinical manifestations of HADHA- and HADHB-related TFP deficiency are similar, the distribution of phenotypes differs. Approximately half of individuals with HADHA pathogenic variants present with a severe/lethal phenotype, while 70% of individuals with HADHB variants have a milder phenotype [Spiekerkoetter et al 2004].
Prevalence
The incidence of LCHAD deficiency on NBS data from Australia, Germany, and the US was estimated at 1:250,000; TFP deficiency incidence was estimated at 1:750,000 [Lindner et al 2010].
The carrier frequency of the most common HADHA pathogenic variant in individuals of European ancestry (c.1528G>C) is estimated to be 1:173 in Estonia, 1:217 in Poland, and 1:240 in Finland [Joost et al 2012]. To date, this variant has not been reported in the Japanese or Korean populations [Purevsuren et al 2009].
LCHAD deficiency is especially frequent in the Pomerania region of Poland near the Baltic Sea, partly as a result of a high carrier frequency (1:73) of HADHA variant c.1528G>C in individuals of Kashubian ancestry; the prevalence is estimated at 1:16,900 [Piekutowska-Abramczuk et al 2010, Nedoszytko et al 2017].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in HADHA or HADHB.
Differential Diagnosis
Table 3.
Genetic Disorders of Interest in the Differential Diagnosis of LCHAD/TFP Deficiency
In addition to the above-mentioned conditions, mitochondrial respiratory chain disorders should be considered in the differential diagnosis of long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) / trifunctional protein (TFP) deficiency. The common manifestations of LCHAD/TFP deficiency – such as cardiomyopathy, skeletal myopathy, hypotonia, peripheral neuropathy, and retinopathy – are also very commonly seen in mitochondrial respiratory chain disorders. See Primary Mitochondrial Disorders Overview.
Management
A brief outline of treatment recommendations for long-chain fatty acid oxidation defects including long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) / trifunctional protein (TFP) deficiency has been published [Spiekerkoetter et al 2009].
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with LCHAD/TFP deficiency, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with LCHAD/TFP Deficiency
Treatment of Manifestations
Management by multidisciplinary specialists including a metabolic physician / biochemical geneticist, specialist metabolic dietitian, cardiologist, neurologist, ophthalmologist, and developmental pediatrician is recommended.
Table 5.
Treatment of Manifestations in Individuals with LCHAD/TFP Deficiency
Table 6.
Emergency Outpatient Treatment in Individuals with LCHAD/TFP Deficiency
Acute manifestations (e.g., lethargy, encephalopathy, intractable vomiting, seizures, severe myalgia, red-colored urine) often occur in the setting of intercurrent illness and/or inadequate caloric intake as a result of poor appetite or prolonged fasting, and should be managed with generous caloric and intravenous fluid support in a hospital setting. Suspected infection should be identified and treated immediately.
Table 7.
Acute Inpatient Treatment in Individuals with LCHAD/TFP Deficiency
Prevention of Primary Manifestations
Avoidance of fasting and supplementation with medium-chain triglycerides (MCT) or triheptanoin remains the mainstay of treatment. Early diagnosis and strict dietary therapy may prevent or delay the onset or slow the progression of long-term complications [Fletcher et al 2012, Fahnehjelm et al 2016, Immonen et al 2016b, De Biase et al 2017, Fraser et al 2019, Dulz et al 2021, Grünert et al 2021].
Education of parents and caregivers is critical to ensure diligent observation and timely initiation of treatment in the setting of intercurrent illness or other catabolic stressors. Prompt administration of high dextrose-containing intravenous fluids is essential to avoid complications such as hypoglycemia, liver failure, rhabdomyolysis, encephalopathy, and coma.
Written protocols for emergency treatment (see Table 8) should be provided to parents, primary care providers / pediatricians, teachers, and school staff. Emergency letters should summarize key information and principles of emergency treatment for LCHAD/TFP deficiency and contain contact information for the primary treating metabolic center. For any planned travel or vacations, consider contacting a center of expertise near the destination prior to travel dates.
Table 8.
Sample Emergency Management Protocol for Individuals with LCHAD/TFP Deficiency
Surveillance
There are no current published guidelines for surveillance. In addition to regular evaluations by a metabolic specialist and metabolic dietician, the evaluations in Table 9 are recommended.
Table 9.
Recommended Surveillance for Individuals with LCHAD/TFP Deficiency
Agents/Circumstances to Avoid
Avoid the following:
- Fasting, including periods of preparation and recovery from planned surgery or anesthesia
- Inadequate caloric provision during stressors, especially when fasting is involved (surgery or procedure requiring fasting/anesthesia)
- Inadequate calories following vaccinationNote: Vaccination is safe.
- Dehydration (risk for rhabdomyolysis and acute renal failure)
- High-fat diet including ketogenic or carbohydrate-restricted diets for the purpose of weight loss, such as Atkins diet
- Administration of intravenous intralipids during an acute metabolic crisis
Anesthetics that contain high doses of long-chain fatty acids (e.g., propofol, etomidate) are avoided in long-chain fatty acid oxidation defects. However, a retrospective analysis revealed no adverse events with propofol for short-duration procedures in individuals with LCHAD/TFP deficiency [Martin et al 2014]. A combination of midazolam, thiopental, fentanyl, and remifentanil was used successfully in an individual with LCHAD deficiency [Steinmann et al 2010].
Evaluation of Relatives at Risk
At-risk sibs of any age. Testing of all at-risk sibs of any age is warranted to identify as early as possible those who would benefit from institution of treatment and preventive measures (see Management, Prevention of Primary Manifestations).
- If the pathogenic variants in the family are known, molecular genetic testing can be used to clarify the genetic status of at-risk sibs.
- If the pathogenic variants in the family are not known, obtain a plasma acylcarnitine profile, plasma free and total carnitine, and urine organic acid profile.
Newborn sibs. For at-risk newborn sibs when prenatal testing was not performed: in parallel with newborn screening (NBS)* either test for the familial HADHA or HADHB pathogenic variants or obtain a plasma acylcarnitine profile, plasma free and total carnitine, and urine organic acid profile.
* The following medical interventions need to begin immediately on receipt of an abnormal NBS result while additional testing is performed to determine whether the abnormal screen represents a true positive NBS result and to establish a definitive diagnosis of LCHAD/TFP deficiency:
- Evaluation of the newborn to ascertain clinical status
- Education of the caregivers to avoid prolonged fasting and to monitor for decreased oral intake, vomiting, or lethargy
- Immediate intervention (to be considered if the newborn is not doing well clinically) possibly including admission to the hospital, fluid resuscitation, infusion of IV dextrose (≥10%), and cardiac evaluation
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Labor and postpartum periods are catabolic states and place the mother at higher risk for rhabdomyolysis and myoglobinuria. A successful pregnancy in a female with LCHAD deficiency has been reported [van Eerd et al 2017]. Management included increasing MCT intake in the third trimester and high dextrose infusion in the peripartum period.
Maternal complications such as HELLP syndrome and acute fatty liver of pregnancy are seen in an estimated 15%-25% of pregnancies in women carrying a fetus affected with LCHAD/TFP deficiency [den Boer et al 2002, Spiekerkoetter et al 2003, Karall et al 2015]. Pregnant females who are heterozygous for a HADHA or HADHB pathogenic variant (including suspected carriers) should be monitored for HELLP syndrome and acute fatty liver of pregnancy. Liver function testing should be performed at each prenatal visit during the first two trimesters and more frequently during the third trimester, when the risk for HELLP syndrome and acute fatty liver of pregnancy is the greatest. Management by a team comprising a maternal-fetal medicine specialist and a medical/biochemical geneticist is recommended.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Cardiac transplantation. Favorable outcome after cardiac transplantation in individuals with TFP deficiency has been reported [Bursle et al 2018]. However, it is expected that with timely diagnosis, strict dietary therapy, and MCT or triheptanoin supplementation, cardiac transplantation may not be required.
Bezafibrate is a hypolipidemic drug and an agonist of peroxisome proliferator-activated receptor (PPAR). It increases expression of several enzymes involved in mitochondrial fatty acid oxidation, including TFP [Djouadi et al 2016]. Bezafibrate was reported to have a favorable outcome in two individuals with TFP deficiency [Suyama et al 2020]. Bezafibrate is not available in the United States.
REN001 (Reneo Pharmaceuticals®) is a selective PPAR-δ agonist that increases transcription of genes involved in mitochondrial fatty acid oxidation. It has received orphan drug designation for LCHAD deficiency from the European Medicines Agency.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency and trifunctional protein (TFP) deficiency are inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are presumed to be heterozygous for an HADHA or HADHB pathogenic variant.
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an HADHA or HADHB pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are not at risk of developing LCHAD/TFP deficiency. However, pregnant female carriers may be at risk of developing HELLP syndrome and acute fatty liver of pregnancy if the fetus has LCHAD/TFP deficiency (see Pregnancy Management).
Sibs of a proband
- If both parents are known to be heterozygous for an HADHA or HADHB pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of inheriting neither of the familial pathogenic variants.
- Significant intrafamilial clinical variability may be observed between sibs who inherit the same biallelic HADHA or HADHB pathogenic variants. However, this variability is considered to stem primarily from environmental factors such as diet and the severity of infection triggering metabolic decompensation [Tyni & Pihko 1999, Bursle et al 2018, Wei et al 2020].
- Heterozygotes (carriers) are not at risk of developing LCHAD/TFP deficiency. However, pregnant female carriers may be at risk of developing HELLP syndrome and acute fatty liver of pregnancy if the fetus has LCHAD/TFP deficiency (see Pregnancy Management).
Offspring of a proband. The offspring of an individual with LCHAD/TFP deficiency are obligate heterozygotes (carriers) for a pathogenic variant in HADHA or HADHB.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an HADHA or HADHB pathogenic variant.
Carrier Detection
Molecular genetic carrier testing for at-risk relatives requires prior identification of the HADHA or HADHB pathogenic variants in the family.
Note: Because biochemical analysis is usually normal in carriers, biochemical testing is not reliable for the detection of carriers.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
- Pregnant females who are heterozygous for an HADHA or HADHB pathogenic variant (including suspected carriers) should be monitored for HELLP syndrome and acute fatty liver of pregnancy (see Pregnancy Management).
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the HADHA or HADHB pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- British Inherited Metabolic Disease Group (BIMDG)TEMPLE (Tools Enabling Metabolic Parents LEarning)United Kingdom
- MedlinePlus
- MedlinePlus
- STAR-G (Screening, Technology and Research in Genetics)Email: info@newbornscreening.info
- STAR-G (Screening, Technology and Research in Genetics)Email: info@newbornscreening.info
- FOD Family Support Group (Fatty Oxidation Disorder)Phone: 517-381-1940Email: deb@fodsupport.org; fodgroup@gmail.com
- International Network for Fatty Acid Oxidation Research and Management
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Newborn Screening in Your StateHealth Resources & Services Administration
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Long-Chain Hydroxyacyl-CoA Dehydrogenase Deficiency / Trifunctional Protein Deficiency: Genes and Databases
Table B.
OMIM Entries for Long-Chain Hydroxyacyl-CoA Dehydrogenase Deficiency / Trifunctional Protein Deficiency (View All in OMIM)
Molecular Pathogenesis
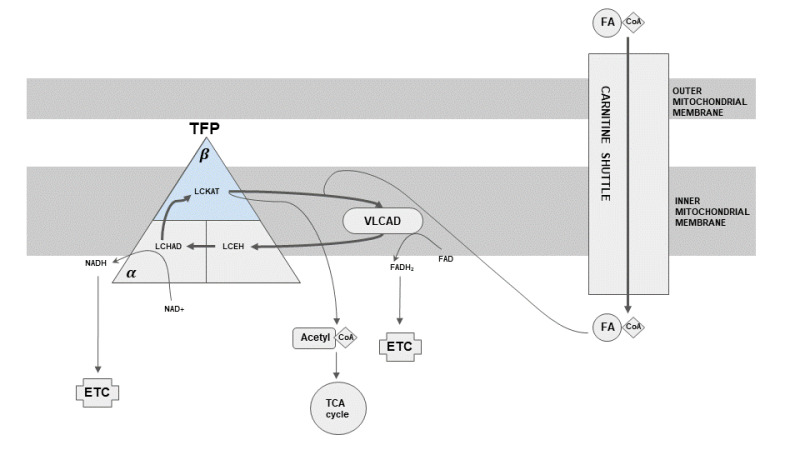
Mitochondrial fatty acid oxidation (beta-oxidation) is the primary pathway of energy production from fatty acids. Fatty acid undergoes repeated cycles of four steps inside mitochondria that result in the shortening of fatty acid by two carbon atoms and production of acetyl-coenzyme A (CoA), reduced nicotinamide adenine dinucleotide (NADH), and reduced flavin adenine dinucleotide (FADH2). Acetyl-CoA can either be utilized to make ketone bodies or oxidized via the tricarboxylic acid cycle for energy generation. High-energy electrons in NADH and FADH2 molecules are transferred to the electron transport chain for ATP generation. The first of these four steps are catalyzed by acyl-CoA dehydrogenase, while the last three are catalyzed by the mitochondrial trifunctional protein (TFP) (see Figure 1).
Mitochondrial TFP is an octamer composed of four alpha subunits (encoded by HADHA) and four beta subunits (encoded by HADHB). The alpha subunit catalyzes long-chain enoyl-CoA hydratase and long-chain 3-hydroxyacyl-CoA dehydrogenase activities, while the beta subunit catalyzes long-chain 3-ketoacyl-CoA thiolase activity.
In addition, the alpha subunit of TFP participates in cardiolipin remodeling, and TFP physically interacts with mitochondrial respiratory chain complex 1 [Taylor et al 2012, Miklas et al 2019, Wang et al 2019]. Resemblance of TFP deficiency to mitochondrial respiratory chain disorders (e.g., elevated lactic acid, cardiomyopathy, polyneuropathy, retinopathy) may be explained by these functional and physical interactions of TFP with the respiratory chain.
Mechanism of disease causation. Loss of function
Table 10.
Notable HADHA Pathogenic Variants
Chapter Notes
Author Notes
Dr Pankaj Prasun is faculty in the Division of Medical Genetics of the Department of Genetics and Genomics at the Icahn School of Medicine at Mount Sinai, New York. He is also the director of the Mitochondrial Medicine Program and has published a textbook on mitochondrial medicine.
PANKAJ PRASUN, MD
Division of Medical Genetics
Department of Genetics and Genomics
Icahn School of Medicine at Mount Sinai
One Gustave L. Levy Place
New York, NY, 10029, USA
Email: moc.liamg@nusarpjaknaprd
Revision History
- 1 September 2022 (sw) Review posted live
- 16 December 2021 (pp) Original submission
References
Literature Cited
- Aradhya S, Lewis R, Bonaga T, Nwokekeh N, Stafford A, Boggs B, Hruska K, Smaoui N, Compton JG, Richard G, Suchy S. Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet Med. 2012;14:594–603. [PubMed: 22382802]
- Blish KR, Ibdah JA. Maternal heterozygosity for a mitochondrial trifunctional protein mutation as a cause for liver disease in pregnancy. Med Hypotheses. 2005;64:96–100. [PubMed: 15533621]
- Bo R, Yamada K, Kobayashi H, Jamiyan P, Hasegawa Y, Taketani T, Fukuda S, Hata I, Niida Y, Shigematsu Y, Iijima K, Yamaguchi S. Clinical and molecular investigation of 14 Japanese patients with complete TFP deficiency: a comparison with Caucasian cases. J Hum Genet. 2017;62:809–14. [PubMed: 28515471]
- Bosch X, Poch E, Grau JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361:62–72. [PubMed: 19571284]
- Boutron A, Acquaviva C, Vianey-Saban C, de Lonlay P, de Baulny HO, Guffon N, Dobbelaere D, Feillet F, Labarthe F, Lamireau D, Cano A, de Villemeur TB, Munnich A, Saudubray JM, Rabier D, Rigal O, Brivet M. Comprehensive cDNA study and quantitative analysis of mutant HADHA and HADHB transcripts in a French cohort of 52 patients with mitochondrial trifunctional protein deficiency. Mol Genet Metab. 2011;103:341–8. [PubMed: 21549624]
- Bursle C, Weintraub R, Ward C, Justo R, Cardinal J, Coman D. Mitochondrial trifunctional protein deficiency: severe cardiomyopathy and cardiac transplantation. JIMD Rep. 2018;40:91–5. [PMC free article: PMC6122028] [PubMed: 29124685]
- De Biase I, Viau KS, Liu A, Yuzyuk T, Botto LD, Pasquali M, Longo N. Diagnosis, treatment, and clinical outcome of patients with mitochondrial trifunctional protein/long-chain 3-hydroxy acyl-CoA dehydrogenase deficiency. JIMD Rep. 2017;31:63–71. [PMC free article: PMC5388644] [PubMed: 27117294]
- den Boer ME, Dionisi-Vici C, Chakrapani A, van Thuijl AO, Wanders RJ, Wijburg FA. Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J Pediatr. 2003;142:684–9. [PubMed: 12838198]
- den Boer ME, Wanders RJ, Morris AA, Ijlst L, Heymans HS, Wijburg FA. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics. 2002;109:99–104. [PubMed: 11773547]
- Diekman EF, Boelen CC, Prinsen BH, Ijlst L, Duran M, de Koning TJ, Waterham HR, Wanders RJ, Wijburg FA, Visser G. Necrotizing enterocolitis and respiratory distress syndrome as first clinical presentation of mitochondrial trifunctional protein deficiency. JIMD Rep. 2013;7:1–6. [PMC free article: PMC3575038] [PubMed: 23430487]
- Djouadi F, Habarou F, Le Bachelier C, Ferdinandusse S, Schlemmer D, Benoist JF, Boutron A, Andresen BS, Visser G, de Lonlay P, Olpin S, Fukao T, Yamaguchi S, Strauss AW, Wanders RJ, Bastin J. Mitochondrial trifunctional protein deficiency in human cultured fibroblasts: effects of bezafibrate. J Inherit Metab Dis. 2016;39:47–58. [PubMed: 26109258]
- Dulz S, Atiskova Y, Engel P, Wildner J, Tsiakas K, Santer R. Retained visual function in a subset of patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). Ophthalmic Genet. 2021;42:23–27. [PubMed: 33107778]
- Elizondo G, Matern D, Vockley J, Harding CO, Gillingham MB. Effects of fasting, feeding and exercise on plasma acylcarnitines among subjects with CPT2D, VLCADD and LCHADD/TFPD. Mol Genet Metab. 2020;131:90–7. [PMC free article: PMC8048763] [PubMed: 32928639]
- Fahnehjelm KT, Liu Y, Olsson D, Amrén U, Haglind CB, Holmström G, Halldin M, Andreasson S, Nordenström A. Most patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency develop pathological or subnormal retinal function. Acta Paediatr. 2016;105:1451–60. [PubMed: 27461099]
- Fletcher AL, Pennesi ME, Harding CO, Weleber RG, Gillingham MB. Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Mol Genet Metab. 2012;106:18–24. [PMC free article: PMC3506186] [PubMed: 22459206]
- Fraser H, Geppert J, Johnson R, Johnson S, Connock M, Clarke A, Taylor-Phillips S, Stinton C. Evaluation of earlier versus later dietary management in long-chain 3-hydroxyacyl-CoA dehydrogenase or mitochondrial trifunctional protein deficiency: a systematic review. Orphanet J Rare Dis. 2019;14:258. [PMC free article: PMC6858661] [PubMed: 31730477]
- Gillingham MB, Heitner SB, Martin J, Rose S, Goldstein A, El-Gharbawy AH, Deward S, Lasarev MR, Pollaro J, DeLany JP, Burchill LJ, Goodpaster B, Shoemaker J, Matern D, Harding CO, Vockley J. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2017;40:831–43. [PMC free article: PMC6545116] [PubMed: 28871440]
- Grünert SC, Eckenweiler M, Haas D, Lindner M, Tsiakas K, Santer R, Tucci S, Spiekerkoetter U. The spectrum of peripheral neuropathy in disorders of the mitochondrial trifunctional protein. J Inherit Metab Dis. 2021;44:893–902. [PubMed: 33638202]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Ijlst L, Ruiter JP, Hoovers JM, Jakobs ME, Wanders RJ. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest. 1996;98:1028–33. [PMC free article: PMC507519] [PubMed: 8770876]
- Immonen T, Ahola E, Toppila J, Lapatto R, Tyni T, Lauronen L. Peripheral neuropathy in patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency - a follow-up EMG study of 12 patients. Eur J Paediatr Neurol. 2016a;20:38–44. [PubMed: 26653362]
- Immonen T, Turanlahti M, Paganus A, Keskinen P, Tyni T, Lapatto R. Earlier diagnosis and strict diets improve the survival rate and clinical course of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr. 2016b;105:549–54. [PubMed: 26676313]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Joost K, Ounap K, Zordania R, Uudelepp ML, Olsen RK, Kall K, Kilk K, Soomets U, Kahre T. Prevalence of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency in Estonia. JIMD Rep. 2012;2:79–85. [PMC free article: PMC3509831] [PubMed: 23430857]
- Karall D, Brunner-Krainz M, Kogelnig K, Konstantopoulou V, Maier EM, Möslinger D, Plecko B, Sperl W, Volkmar B, Scholl-Bürgi S. Clinical outcome, biochemical and therapeutic follow-up in 14 Austrian patients with long-chain 3-hydroxy acyl CoA dehydrogenase deficiency (LCHADD). Orphanet J Rare Dis. 2015;10:21. [PMC free article: PMC4407779] [PubMed: 25888220]
- Kobayashi T, Minami S, Mitani A, Tanizaki Y, Booka M, Okutani T, Yamaguchi S, Ino K. Acute fatty liver of pregnancy associated with fetal mitochondrial trifunctional protein deficiency. J Obstet Gynaecol Res. 2015;41:799–802. [PubMed: 25420603]
- Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33:521–6. [PubMed: 20373143]
- Martin JM, Gillingham MB, Harding CO. Use of propofol for short duration procedures in children with long chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiencies. Mol Genet Metab. 2014;112:139–42. [PMC free article: PMC4121654] [PubMed: 24780638]
- Miklas JW, Clark E, Levy S, Detraux D, Leonard A, Beussman K, Showalter MR, Smith AT, Hofsteen P, Yang X, Macadangdang J, Manninen T, Raftery D, Madan A, Suomalainen A, Kim DH, Murry CE, Fiehn O, Sniadecki NJ, Wang Y, Ruohola-Baker H. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat Commun. 2019;10:4671. [PMC free article: PMC6789043] [PubMed: 31604922]
- Nedoszytko B, Siemińska A, Strapagiel D, Dąbrowski S, Słomka M, Sobalska-Kwapis M, Marciniak B, Wierzba J, Skokowski J, Fijałkowski M, Nowicki R, Kalinowski L. High prevalence of carriers of variant c.1528G>C of HADHA gene causing long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD) in the population of adult Kashubians from North Poland. PLoS One. 2017;12:e0187365. [PMC free article: PMC5667839] [PubMed: 29095929]
- Okun JG, Kölker S, Schulze A, Kohlmüller D, Olgemöller K, Lindner M, Hoffmann GF, Wanders RJ, Mayatepek E. A method for quantitative acylcarnitine profiling in human skin fibroblasts using unlabelled palmitic acid: diagnosis of fatty acid oxidation disorders and differentiation between biochemical phenotypes of MCAD deficiency. Biochim Biophys Acta. 2002;1584:91–8. [PubMed: 12385891]
- Piekutowska-Abramczuk D, Olsen RK, Wierzba J, Popowska E, Jurkiewicz D, Ciara E, Ołtarzewski M, Gradowska W, Sykut-Cegielska J, Krajewska-Walasek M, Andresen BS, Gregersen N, Pronicka E. A comprehensive HADHA c.1528G>C frequency study reveals high prevalence of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency in Poland. J Inherit Metab Dis. 2010;33 Suppl 3:S373–7. [PubMed: 20814823]
- Purevsuren J, Fukao T, Hasegawa Y, Kobayashi H, Li H, Mushimoto Y, Fukuda S, Yamaguchi S. Clinical and molecular aspects of Japanese patients with mitochondrial trifunctional protein deficiency. Mol Genet Metab. 2009;98:372–7. [PubMed: 19699128]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Roe CR, Brunengraber H. Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: review of 15 years experience. Mol Genet Metab. 2015;116:260–8. [PMC free article: PMC4712637] [PubMed: 26547562]
- Sklirou E, Alodaib AN, Dobrowolski SF, Mohsen AA, Vockley J. Physiological perspectives on the use of triheptanoin as anaplerotic therapy for long chain fatty acid oxidation disorders. Front Genet. 2021;11:598760. [PMC free article: PMC7875087] [PubMed: 33584796]
- Spiekerkoetter U, Khuchua Z, Yue Z, Bennett MJ, Strauss AW. General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatr Res. 2004;55:190–6. [PubMed: 14630990]
- Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, Das A, Haase C, Hennermann JB, Karall D, de Klerk H, Knerr I, Koch HG, Plecko B, Röschinger W, Schwab KO, Scheible D, Wijburg FA, Zschocke J, Mayatepek E, Wendel U. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009;32:498–505. [PubMed: 19452263]
- Spiekerkoetter U, Sun B, Khuchua Z, Bennett MJ, Strauss AW. Molecular and phenotypic heterogeneity in mitochondrial trifunctional protein deficiency due to beta-subunit mutations. Hum Mutat. 2003;21:598–607. [PubMed: 12754706]
- Steinmann D, Knab J, Priebe HJ. Perioperative management of a child with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Paediatr Anaesth. 2010;20:371–3. [PubMed: 20470346]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Suyama T, Shimura M, Fushimi T, Kuranobu N, Ichimoto K, Matsunaga A, Takayanagi M, Murayama K. Efficacy of bezafibrate in two patients with mitochondrial trifunctional protein deficiency. Mol Genet Metab Rep. 2020;24:100610. [PMC free article: PMC7264074] [PubMed: 32509533]
- Szugye HS. Pediatric rhabdomyolysis. Pediatr Rev. 2020;41:265–75. [PubMed: 32482689]
- Taylor WA, Mejia EM, Mitchell RW, Choy PC, Sparagna GC, Hatch GM. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS One. 2012;7:e48628. [PMC free article: PMC3494688] [PubMed: 23152787]
- Tyni T, Kivelä T, Lappi M, Summanen P, Nikoskelainen E, Pihko H. Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: a new type of hereditary metabolic chorioretinopathy. Ophthalmology. 1998;105:810–24. [PubMed: 9593380]
- Tyni T, Pihko H. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr. 1999;88:237–45. [PubMed: 10229030]
- Tyni T, Rapola J, Palotie A, Pihko H. Hypoparathyroidism in a patient with long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency caused by the G1528C mutation. J Pediatr. 1997;131:766–8. [PubMed: 9403664]
- van Eerd DC, Brussé IA, Adriaens VF, Mankowski RT, Praet SF, Michels M, Langeveld M. Management of an LCHADD patient during pregnancy and high intensity exercise. JIMD Rep. 2017;32:95–100. [PMC free article: PMC5355378] [PubMed: 27334895]
- van Vliet P, Berden AE, van Schie MKM, Bakker JA, Heringhaus C, de Coo IFM, Langeveld M, Schroijen MA, Arbous MS. Peripheral neuropathy, episodic rhabdomyolysis, and hypoparathyroidism in a patient with mitochondrial trifunctional protein deficiency. JIMD Rep. 2018;38:101–105. [PMC free article: PMC5874207] [PubMed: 28685493]
- Vockley J, Burton B, Berry G, Longo N, Phillips J, Sanchez-Valle A, Chapman K, Tanpaiboon P, Grunewald S, Murphy E, Lu X, Cataldo J. Effects of triheptanoin (UX007) in patients with long-chain fatty acid oxidation disorders: Results from an open-label, long-term extension study. J Inherit Metab Dis. 2021;44:253–63. [PMC free article: PMC7891391] [PubMed: 32885845]
- Vockley J, Charrow J, Ganesh J, Eswara M, Diaz GA, McCracken E, Conway R, Enns GM, Starr J, Wang R, Abdenur JE, Sanchez-de-Toledo J, Marsden DL. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol Genet Metab. 2016;119:223–31. [PMC free article: PMC5083220] [PubMed: 27590926]
- Wang J, Zhan H, Li FY, Pursley AN, Schmitt ES, Wong LJ. Targeted array CGH as a valuable molecular diagnostic approach: experience in the diagnosis of mitochondrial and metabolic disorders. Mol Genet Metab. 2012;106:221–30. [PubMed: 22494545]
- Wang Y, Palmfeldt J, Gregersen N, Makhov AM, Conway JF, Wang M, McCalley SP, Basu S, Alharbi H, St Croix C, Calderon MJ, Watkins S, Vockley J. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J Biol Chem. 2019;294:12380–91. [PMC free article: PMC6699831] [PubMed: 31235473]
- Wei CJ, Chang XZ, Ge L, Fu XN, Fan YB, Liu JY, Wang S, Li HL, Yang YL, Xiong H. Multisystem involvement in Chinese patients with neuromyopathic phenotype of mitochondrial trifunctional protein deficiency. Chin Med J (Engl). 2020;133:1358–60. [PMC free article: PMC7289308] [PubMed: 32515919]
- Zöggeler T, Stock K, Jörg-Streller M, Spenger J, Konstantopoulou V, Hufgard-Leitner M, Scholl-Bürgi S, Karall D. Long-term experience with triheptanoin in 12 Austrian patients with long-chain fatty acid oxidation disorders. Orphanet J Rare Dis. 2021;16:28. [PMC free article: PMC7807521] [PubMed: 33446227]
Publication Details
Author Information and Affiliations
Icahn School of Medicine at Mount Sinai
New York, New York
Icahn School of Medicine at Mount Sinai
New York, New York
Icahn School of Medicine at Mount Sinai
New York, New York
Publication History
Initial Posting: September 1, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Prasun P, LoPiccolo MK, Ginevic I. Long-Chain Hydroxyacyl-CoA Dehydrogenase Deficiency / Trifunctional Protein Deficiency. 2022 Sep 1. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.