Clinical Description
Mitochondrial DNA-associated Leigh syndrome spectrum (mtDNA-LSS) is a continuum of progressive neurodegenerative disorders, with typical onset in infancy or early childhood of sudden neurodevelopmental regression. Individuals with LSS may have variable extraneurologic manifestations including gastrointestinal, cardiac, hepatic, renal, and endocrine disease. The clinical features of mtDNA-LSS are similar to those observed in individuals with nuclear gene-encoded Leigh syndrome spectrum (LSS). Clinically, it is difficult to distinguish mtDNA-LSS from nuclear gene-encoded LSS, and there are relatively few publications describing the full range of clinical features in mtDNA-LSS. The following description of the phenotypic features summarizes the more robust data on features of all individuals with LSS; specific information and references for mtDNA-LSS are included if available.
Table 2.
Mitochondrial DNA-Associated Leigh Syndrome Spectrum: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
| LSS 1 | mtDNA-LSS 2 |
|---|
|
Neurologic features
|
Developmental delay
| >70% | >75% | |
|
Regression
| ~50% | Unknown 3 | |
|
Hypotonia
| ~60% | ~65% | |
|
Dystonia
| ~35% | ~30% | |
|
Ataxia
| ~30% | ~50% | |
|
Spasticity
| ~40% | ~50% | |
|
Muscle weakness
| ~30% | ~75% | |
|
Peripheral neuropathy
| ~10% | ~5% | |
|
Dysphagia
| ~25% | ~80% | |
|
Epilepsy
| ~30% | ~40% | |
|
Respiratory abnormalities
| ~30% | ~35% | Apnea, hypoventilation, hyperventilation, irregular respiration |
|
Sensorineural hearing loss
| 15% | 15% | |
|
Ophthalmologic features
|
Ophthalmoplegia
| ~25% | ~30% | |
|
Optic atrophy
| ~15% | ~20% | |
|
Retinopathy
| ~10% | <10% | |
|
Other
|
Poor weight gain
| ~40% | ~30% | |
|
Gastrointestinal manifestations
| ~25% | ~25% | Vomiting, gastroparesis, intestinal dysmotility, constipation |
|
Cardiac manifestations
| ~15% | ~20% | Hypertrophic or dilated cardiomyopathy, conduction defects |
|
Hepatic manifestations
| <10% | ~15% | ↑ liver transaminases, hepatomegaly, liver failure |
|
Renal manifestations
| <5% | <5% | Tubulopathy |
|
Endocrine manifestations
| <5% | <5% | Diabetes, adrenal insufficiency |
mtDNA = mitochondrial DNA; LSS = Leigh syndrome spectrum
- 1.
Estimated percentages are based on several cohorts of individuals with nuclear gene-encoded LSS and mtDNA-LSS (~1,000 individuals); however, each clinical feature was not reported for every individual [Rahman et al 1996, Naess et al 2009, Sofou et al 2014, Sofou et al 2018, Wei et al 2018, Alves et al 2020, Hong et al 2020, Lee et al 2020, Ardissone et al 2021, Lim et al 2022, Stenton et al 2022, Ardissone et al 2023, Kistol et al 2023].
- 2.
- 3.
Data not available for regression, as this was often described in conjunction with developmental delay.
Onset of symptoms is typically between age three and 12 months, often following an intercurrent illness (usually viral) or metabolic challenge (vaccinations, surgery, prolonged fasting). However, disease onset can vary, ranging from prenatal onset to adulthood. Early onset of symptoms within the first 24 months of life has been reported in approximately 75% of individuals with LSS. Later onset (age >2 years) includes presentation in childhood, adolescence, or adulthood and is generally associated with slower progression of disease.
Initial features may be nonspecific and vary with age of onset:
Early-onset LSS (age <2 years) may present with nonspecific features such as feeding difficulties, poor weight gain, hypotonia, developmental delay, and persistent vomiting.
Late-onset LSS (age >2 years) may manifest with predominant muscle weakness and movement disorders including ataxia and dystonia.
Decompensation (often with increased blood and/or cerebrospinal fluid [CSF] lactate concentrations) during an intercurrent illness is typically associated with sudden loss of developmental skills. A period of recovery may follow the initial decompensation, but the individual rarely returns to the developmental status achieved prior to the presenting illness. Most individuals with LSS will experience at least one acute decompensation that may require hospitalization.
Neurologic features include developmental delay/regression, hypotonia, spasticity, dystonia, muscle weakness, hypo- or hyperreflexia, seizures (myoclonic, generalized tonic-clonic, infantile spasms), movement disorders (including chorea), cerebellar ataxia, and peripheral neuropathy. Adolescents and adults with LSS may exhibit psychiatric disturbance.
Brain stem lesions may cause respiratory difficulty (apnea, central hypo- or hyperventilation, or irregular respiration), bulbar problems (e.g., abnormal swallowing and speech), persistent vomiting, and abnormalities of thermoregulation (hypo- and hyperthermia). Sensorineural hearing loss may be present in approximately 15% of individuals, originating from the cochlea or auditory nerves.
Ophthalmologic findings include optic atrophy, retinitis pigmentosa, strabismus, ptosis, nystagmus, and ophthalmoparesis. Pigmentary retinopathy was reported in 12 out of 13 individuals with an MT-ATP6
m.8993T>G pathogenic variant in one retrospective cohort [Ng et al 2019].
Gastrointestinal manifestations include poor weight gain, vomiting, dysphagia, gastroparesis, and intestinal dysmotility. One natural history study suggested that gastrointestinal symptoms were the most common extraneurologic manifestation in individuals with LSS, with 25% exhibiting gut dysmotility and nearly half of the cohort requiring gastrostomy or nasogastric tube feeding at follow up [Lim et al 2022].
Cardiac manifestations including hypertrophic or dilated cardiomyopathy and conduction defects (e.g., Wolff-Parkinson-White syndrome and paroxysmal supraventricular tachycardia) are reported in approximately 15% of individuals with LSS. Cardiomyopathy has been reported in up to 70% of individuals with an MT-ND5 pathogenic variant [Stenton et al 2022, Kistol et al 2023].
Hepatic manifestations including elevated liver transaminases, hepatomegaly, or liver failure have been reported in approximately 10% of individuals with LSS [Naess et al 2009, Van Hove et al 2010, Sofou et al 2014, Duff et al 2015, Alves et al 2020].
Renal manifestations including renal tubulopathy or diffuse glomerulocystic kidney damage have been reported in approximately 5% of individuals with LSS [López et al 2006, Naess et al 2009, Sofou et al 2018, Alves et al 2020, Hong et al 2020, Stenton et al 2022].
Endocrine manifestations including diabetes mellitus and adrenal insufficiency have been reported in a small proportion of individuals with LSS [Alves et al 2020, Ardissone et al 2023].
Prognosis in individuals with LSS is generally poor. Most affected individuals experience episodic deterioration of motor and/or cognitive function interspersed with plateaus in development. Up to 50% of individuals with LSS will die before age three years, most often from respiratory or cardiac failure, neurologic deterioration, or sepsis. However, disease progression can be variable. Some individuals have slower progression of disease, with stable periods lasting years in between decompensations, even into adulthood. In previously undiagnosed individuals, death may appear to be sudden and unexpected.
Poor prognosis has been associated with early-onset disease [Sofou et al 2014, Lee et al 2016, Hong et al 2020, Ogawa et al 2020]. Individuals with pathogenic variants in MT-ND5 [Ogawa et al 2020] and those with MT-ATP6 pathogenic variant m.8993T>G [Stendel et al 2020, Na & Lee 2022] have been reported to be more likely to have a severe disease phenotype.
Intermediate phenotypes. Maternal relatives of individuals with mtDNA-LSS can have any one or a combination of clinical manifestations of mtDNA-LSS, NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa), or other mitochondrial disorders. These include mild learning difficulties, ataxia, muscle weakness, night blindness, deafness, diabetes mellitus, migraine, or sudden unexpected death (see Genetically Related Disorders).
Genotype-Phenotype Correlations
For most mtDNA pathogenic variants, it is difficult to distinguish a correlation between genotype and phenotype because clinical expression of mtDNA pathogenic variants is influenced by pathogenicity of the variant, relative amount of abnormal and wild type mtDNA (heteroplasmy level), the variation in heteroplasmy level in different tissues, and the energy requirements of brain and other tissues, which may vary with age. Intrafamilial and interfamilial clinical variability are seen in individuals with the same genotype [Stendel et al 2020].
Several studies have tried to describe genotype-phenotype correlations in mtDNA-LSS; however, these are generally inconsistent between cohorts, limiting their translation into clinical practice.
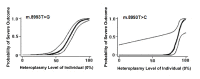
MT-ATP6 pathogenic variants, specifically m.8993T>G and m.8993T>C, probably show the strongest genotype-phenotype correlation of any mtDNA pathogenic variants. They show very little tissue-dependent [Ng et al 2019] or age-dependent variation in the proportion of abnormal mtDNA [White et al 1999b] as well as a strong correlation between the heteroplasmy level and disease severity. Logistic regression models were developed that predict the probability of a severe outcome in an individual based on the measured heteroplasmy level of m.8993T>G and m.8993T>C (see ) [White et al 1999a]. Across all MT-ATP6 variants, heteroplasmy levels have been shown to have a significant negative association with age of onset of disease [Ganetzky et al 2019]. These findings are supported by Ng et al [2019], who generated logistic regression models to predict the risk of disease manifestation based on the blood heteroplasmy level of MT-ATP6 pathogenic variants m.8993T>C, m.8993T>G, m.9176T>C, and m.9185T>C (see ). Note, however, that in such retrospective studies it is not possible to completely avoid ascertainment bias, and the data should be regarded as broadly indicative rather than precise.
Estimated probability of a severe outcome (95% CI) for an individual with MT-ATP6 pathogenic variants m.8993T>G or m.8993T>C, based on the heteroplasmy level (i.e., proportion of abnormal mitochondrial DNA) in the individual. A severe (more...)
Estimated probability of having clinical manifestations of Leigh syndrome spectrum based on the heteroplasmy level detected in blood for MT-ATP6 pathogenic variants m.8993T>C, m.8993T>G, m.9176T>C, and m.9185T>C [Ng et (more...)
MT-ATP6 pathogenic variants m.8993T>G and m.8993T>C tend to be associated with early-onset and severe clinical disease, with m.8993T>G causing a more severe phenotype [Sofou et al 2018, Ganetzky et al 2019, Stendel et al 2020]. Although the cause of this difference is unknown, it is postulated to result from the instability of the abnormal protein.
m.8993T>G. Individuals with a heteroplasmy level below 60% are usually asymptomatic or have only mild pigmentary retinopathy or migraine headaches; however, asymptomatic adults with up to 75% abnormal mtDNA have been reported [
Ganetzky et al 2019,
Ng et al 2019,
Stendel et al 2020]. Individuals with moderate heteroplasmy levels (70%-90%) of pathogenic variant m.8993T>G may present with the milder phenotype of NARP, while those with more than 90% abnormal mtDNA typically present with LSS [
Ng et al 2019,
Stendel et al 2020,
Stenton et al 2022].
m.8993T>C is a less severe variant than m.8993T>G, and generally all symptomatic individuals with m.8993T>C have a heteroplasmy level greater than 90%. However, some asymptomatic individuals with m.8993T>C have also been reported to have a heteroplasmy level greater than 90% [
Stendel et al 2020,
Stenton et al 2022].
Note: Overlap in
MT-ATP6 pathogenic variant heteroplasmy level is observed between some asymptomatic individuals and others with NARP or mild neurologic manifestations, particularly for variants other than m.8993T>G. There can also be overlap in heteroplasmy levels between some individuals with NARP and others with LSS [
Claeys et al 2016,
Ganetzky et al 2019,
Ng et al 2019,
Stendel et al 2020].
Genotype-phenotype correlations are much weaker for other mtDNA pathogenic variants (e.g., m.3243A>G in MT-TL1, m.8344A>G in MT-TK, m.9176T>C in MT-ATP6, m.14459G>A and m.14487T>C in MT-ND6, m.10158T>C and m.10191T>C in MT-ND3, and m.13513G>A in MT-ND5). It is not possible to use heteroplasmy level to predict outcome (e.g., in asymptomatic family members or in prenatal diagnosis) unless the value is near 0% or near 100%. Some individuals with MT-ND3 and MT-ND5 pathogenic variants have been reported to have relatively severe phenotypes at low heteroplasmy levels [Wei et al 2018, Ogawa et al 2020, Stenton et al 2022, Na & Lee 2023].
Other genotype-phenotype correlations have been suggested for mtDNA pathogenic variants affecting respiratory chain enzyme complex I, although confirmation of these findings requires further systematic analysis of larger cohorts. Individuals with complex I deficiency are more commonly reported to have cardiac and visual involvement [Sofou et al 2018], with pathogenic variants in MT-ND5 strongly associated with cardiomyopathy [Stenton et al 2022, Kistol et al 2023]. A retrospective study of neuroimaging in children with mtDNA-LSS suggested that cerebral cortical and white matter involvement is more common in MT-ND5- and MT-ND3-related LSS [Alves et al 2020]; however, this finding was not supported by a subsequent study [Stenton et al 2022]. Poor prognosis has been associated with MT-ND5 pathogenic variants in addition to MT-ATP6 pathogenic variant m.8993T>G [Ogawa et al 2020, Stenton et al 2022].
Individuals with mtDNA-LSS may exhibit overlapping phenotypes with other mitochondrial phenotypes, including MELAS. MT-ND pathogenic variants (most commonly involving MT-ND5 and MT-ND3) have been described in multiple individuals with an LSS/MELAS overlapping phenotype in which individuals display clinical and radiologic features of both syndromes [Wei et al 2021].
Nomenclature
Leigh syndrome was originally described in an affected infant as "subacute necrotizing encephalomyelopathy" [Leigh 1951]. Leigh syndrome was historically defined by characteristic neuropathologic features including multiple focal symmetric necrotic lesions in the basal ganglia, thalamus, brain stem, dentate nuclei, and optic nerves that histologically demonstrate a spongiform appearance and are characterized by demyelination, gliosis, and vascular proliferation. The advent of MRI and next-generation sequencing has made it possible to establish the diagnosis without postmortem examination. Individuals with mtDNA-LSS are often referred to as having "maternally inherited Leigh syndrome" (MILS) [Ciafaloni et al 1993]. This term can cause confusion given perhaps one quarter of probands with mtDNA-LSS have de novo pathogenic variants (see Genetic Counseling).
The term Leigh-like syndrome was used previously when clinical and other features were strongly suggestive of Leigh syndrome but did not fulfill the stringent diagnostic criteria because of atypical neuropathology (i.e., variation in the distribution or character of lesions or the presence of unusual features such as extensive cortical destruction), neuroimaging inconsistent with Leigh syndrome, normal blood and CSF lactate levels, and/or incomplete evaluation.
Leigh syndrome spectrum encompasses both Leigh syndrome and Leigh-like syndrome. Leigh syndrome and Leigh-like syndrome represent a continuum of overlapping clinical and biochemical features and result from pathogenic variants in the same group of mitochondrial and nuclear genes.
Prevalence
LSS is the most frequently observed phenotype of pediatric-onset mitochondrial disorders. Mitochondrial DNA-associated LSS accounts for approximately 30% of LSS.
The following prevalence data are for all genetic causes of LSS. In southeastern Australia, Leigh syndrome occurs in 1:77,000 infants, and the combined birth prevalence of LSS was estimated to be at least 1:40,000 [Rahman et al 1996]. In western Sweden, the prevalence of Leigh syndrome in preschool children was 1:34,000 [Darin et al 2001]. Thus, the prevalence of LSS is likely to be 1:30,000-40,000.
Analyses of 67 individuals with LSS reported by Rahman et al [1996] identified mtDNA pathogenic variants in 34% [Authors, unpublished data]. Hence, the prevalence of mtDNA-LSS is likely to be 1:100,000-140,000.