Summary
Clinical characteristics.
EZH2-related overgrowth is a variable overgrowth syndrome characterized by tall stature, macrocephaly, variable intellect (ranging from normal intellect to severe intellectual disability), characteristic facial appearance, and a range of associated clinical features including advanced bone age, poor coordination, soft, doughy skin, camptodactyly of the fingers and/or toes, umbilical hernia, abnormal tone, and hoarse, low cry in infancy. Brain MRI has identified abnormalities in a few individuals with EZH2-related overgrowth. Neuroblastoma occurs at a slightly increased frequency in individuals with a heterozygous EZH2 pathogenic variant, but data are insufficient to determine absolute risk. There is currently no evidence that additional malignancies (including hematologic malignancies) occur with increased frequency, though a few have been reported.
Diagnosis/testing.
The diagnosis of EZH2-related overgrowth is based on detection of a heterozygous germline EZH2 pathogenic variant on molecular genetic testing.
Management.
Treatment of manifestations: For individuals with developmental delay and/or learning disability, referral for learning/behavior/speech assessment and support may be indicated. Occasionally, toe camptodactyly may require surgical release. Physiotherapy may be of benefit to those experiencing joint pain secondary to ligamentous laxity or joint contractures. Standard treatment with appropriate specialist referral(s) is indicated for epilepsy, scoliosis, and other clinical issues.
Surveillance: Regular medical follow up of young children with EZH2-related overgrowth to monitor developmental progress, camptodactyly (for resolution/improvement), and/or hypotonia; medical follow up of older children/teenagers who do not have medical complications may be less frequent. If scoliosis is present, monitoring per the recommendations of an orthopedist. Although current data do not support specific tumor surveillance programs, clinicians should have a low threshold for investigating any possible tumor-related symptoms.
Pregnancy management: Families and their health care providers should be made aware that an affected baby may be large so that appropriate delivery plans can be made.
Genetic counseling.
EZH2-related overgrowth is inherited in an autosomal dominant manner. Some individuals diagnosed with EZH2-related overgrowth have an affected parent; some individuals diagnosed with EZH2-related overgrowth have the disorder as the result of a de novo EZH2 pathogenic variant. The proportion of individuals with EZH2-related overgrowth caused by a de novo pathogenic variant is unknown. Each child of an individual with EZH2-related overgrowth has a 50% chance of inheriting the pathogenic variant; the phenotype in individuals who inherit a familial EZH2 pathogenic variant cannot be predicted. Once the pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
GeneReview Scope
The scope of this GeneReview encompasses the broad phenotypic spectrum associated with heterozygous EZH2 pathogenic variants, which ranges from classic EZH2-related Weaver syndrome at one end of the spectrum to tall stature at the other end.
Diagnosis
Suggestive Findings
EZH2-related overgrowth should be suspected in an individual with the following clinical and imaging findings [Tatton-Brown et al 2013, Imagawa et al 2023].
Clinical findings
- Tall stature (height or length ≥2 standard deviations [SD] above the mean)
- Macrocephaly (head circumference ≥2 SD above the mean)
- Intellectual disability
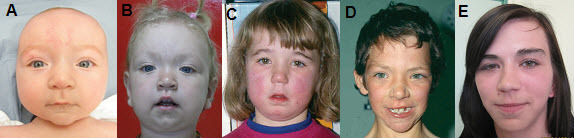
- Characteristic facial appearance (See Figure 1.)
- In children younger than age three years: retrognathia, large, fleshy ears, and a "stuck on" appearance of the chin associated with a horizontal skin crease and sometimes a central dimple
- In affected individuals of all ages, additional features include broad forehead (increased bifrontal diameter), round face, widely spaced eyes, almond-shaped palpebral fissures, and long or prominent philtrum
The characteristic facial appearance (which is most distinctive at a younger age) evolves over time; therefore, review of younger childhood photographs may help the clinician reach a clinical diagnosis. - Poor coordination
- Soft, doughy skin
- Excessive loose skin
- Camptodactyly of the fingers and/or toes (See Note.)
- Umbilical hernia (that is occasionally significant enough to require surgical reduction)
- Abnormal tone (central hypotonia and/or peripheral hypertonia) (See Note.)
- Hoarse, low-pitched cry (sometimes described as a quiet cry)

Figure 1.
Retrognathia present in younger children with EZH2-related overgrowth usually resolves with age. In individuals of all ages the palpebral fissures are frequently almond shaped and the eyes are widely spaced.
Note: A detailed medical history may be necessary to determine if these findings were present in the newborn period / infancy, given that they can resolve or improve throughout childhood.
Imaging findings
- Skeletal imaging. Advanced bone age on plain radiographs or skeletal surveys
- Neuroimaging. The true prevalence of brain MRI abnormalities in EZH2-related overgrowth is currently uncertain, as baseline brain MRI scans are not routinely performed. However, the following brain MRI abnormalities have been reported, although it is currently unclear whether they are associated with EZH2-related disorders or incidental: ventriculomegaly, periventricular leukomalacia, cerebellar infarct, cerebellar hypoplasia, and neuronal migration defects (polymicrogyria) [Gibson et al 2012, Al-Salem et al 2013, Tatton-Brown et al 2013, Cohen et al 2016, Griffiths et al 2019].
Establishing the Diagnosis
The diagnosis of EZH2-related overgrowth is established in a proband by identification of a heterozygous germline EZH2 pathogenic (or likely pathogenic) variant on molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variant" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a heterozygous EZH2 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of EZH2-related overgrowth has not been considered are more likely to be diagnosed using comprehensive genomic testing (see Option 2). Further, epigenetic signature analysis or a methylation array is an option.
Option 1
When the phenotypic findings suggest the diagnosis of EZH2-related overgrowth, molecular genetic testing approaches can include use of a multigene panel.
A multigene panel that includes EZH2 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the diagnosis of EZH2-related overgrowth has not been considered because an individual has atypical phenotypic features, comprehensive genomic testing through exome or genome sequencing does not require the clinician to determine which gene is likely involved. To date, the majority of reported EZH2 pathogenic variants (e.g., missense, nonsense) are within the coding region and are likely to be identified on exome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in EZH2-Related Overgrowth
Epigenetic Signature Analysis / Methylation Array
Pathogenic variants in EZH2 generate a distinctive epigenetic signature that can be used to distinguish between pathogenic and benign variants as well as between loss-of-function and gain-of-function variants and facilitate detection of somatic mosaicism [Choufani et al 2020].
Epigenetic signature analysis of a peripheral blood sample or DNA banked from a blood sample can therefore be considered to clarify the diagnosis in individuals with: (1) suggestive clinical findings of EZH2-related overgrowth but in whom no pathogenic variant in EZH2 has been identified via sequence analysis; or (2) suggestive clinical findings of EZH2-related overgrowth and an EZH2 variant of uncertain clinical significance identified by molecular genetic testing. For an introduction to epigenetic signature analysis click here.
Clinical Characteristics
Clinical Description
Primary phenotypic features associated with EZH2-related overgrowth include tall stature, macrocephaly, and intellectual disability, which are observed in association with a characteristic facial appearance (round face, flattened occiput, hypertelorism, almond-shaped palpebral fissures, retrognathia, large, fleshy ears, and a "stuck on" appearance to the chin) [Tatton-Brown et al 2013, Imagawa et al 2023].
The phenotypic spectrum associated with germline EZH2 pathogenic variants is broad, with classic EZH2-related Weaver syndrome at one end of the spectrum and tall stature at the other. Although most individuals diagnosed with a heterozygous germline EZH2 pathogenic variant have been identified because of a clinical suspicion of Weaver syndrome, a minority have been identified through molecular genetic testing of family members of probands or individuals with overgrowth who did not have a clinical diagnosis of Weaver syndrome [Tatton-Brown et al 2011, Gibson et al 2012]. Thus, the full extent of the phenotypic spectrum of heterozygous EZH2 pathogenic variants is not yet known.
To date, at least 68 individuals have been identified with a pathogenic variant in EZH2 [Tatton-Brown et al 2011, Gibson et al 2012, Al-Salem et al 2013, Tatton-Brown et al 2013, Cohen et al 2016, Usemann et al 2016, Lui et al 2018, Griffiths et al 2019, Turkkahraman et al 2021, Imagawa et al 2023, Oh et al 2023]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
EZH2-Related Overgrowth: Frequency of Select Features
Growth. From data available on 23 newborns, the mean birth length was 2.2 standard deviations (SD) above the mean, with a range of 0.5 SD below the mean to 4.9 SD above the mean. The mean birth weight of 45 newborns was 1.7 SD above the mean, with a range of 1.6 SD below the mean to 4.6 SD above the mean [Tatton-Brown et al 2013].
Tall stature is a near-consistent finding: height in 59 of 65 individuals was at least two SD above the mean (ages 1-70 years). Of note, three of four individuals with a height less than two SD above the mean had been tall as young children. The mean postnatal height was 3.5 SD above the mean.
Of 59 individuals for whom information is available, 27 had a head circumference less than two SD above the mean and 32 had macrocephaly (>2 SD above the mean), with a head circumference reaching up to 5.5 SD above the mean. Head circumferences at birth were at least two SD above the mean in 5 of 6 infants for whom data is available [Gibson et al 2012, Cohen et al 2016]. These data suggest that macrocephaly in EZH2-related overgrowth is likely to be present from birth but longitudinal follow up is required.
Neurologic. Ventriculomegaly, reported in seven individuals, was generally associated with normal cerebrospinal fluid pressure and did not require shunting [Gibson et al 2012, Tatton-Brown et al 2013, Griffiths et al 2019]. Other reported brain MRI findings include neuronal migration defects (including polymicrogyria in several individuals), periventricular leukomalacia (two individuals), and cerebellar abnormalities (two individuals).
Intellectual disability in those with a brain MRI abnormality was:
- Mild in seven individuals (ventriculomegaly [5 individuals], periventricular leukomalacia [1 individual], and cerebellar hypoplasia [1 individual]);
- Moderate in three individuals (periventricular leukomalacia with ventriculomegaly [1 individual] and isolated ventriculomegaly [2 individuals]);
- Severe in two individuals with polymicrogyria and pachygyria [Tatton-Brown et al 2013, Griffiths et al 2019]; in contrast, the individual with polymicrogyria reported by Al-Salem et al [2013] had normal developmental milestones and body asymmetry (left side smaller than the right) with brisk reflexes and increased tone on the left. The degree of intellectual disability of the individual with polymicrogyria reported by Cohen et al [2016] is unknown.Note: The degree of intellectual disability was not reported for one individual with ventriculomegaly.
Several reported individuals with EZH2-related overgrowth had seizures. Four individuals had afebrile seizures [Gibson et al 2012, Usemann et al 2016, Griffiths et al 2019]. Seizure types include tonic-clonic (age of onset 13 years) [1 individual] and brief absence (age of onset 15 years) [Gibson et al 2012]. Two individuals had seizures associated with a febrile illness [Tatton-Brown et al 2013, Cohen et al 2016].
Abnormal tone. In general, abnormal tone (hypotonia, hypertonia, or mixed central hypotonia and peripheral hypertonia), if present, resolves during childhood.
- Hypotonia (predominantly central) was reported in 22 of 47 individuals.
- Hypertonia (predominantly peripheral manifesting as stiffness in the limbs with brisk reflexes) was reported in 14 of 51 individuals.Note: Three of the individuals presenting with peripheral hypertonia were also reported to have central hypotonia [Tatton-Brown et al 2013].
Cognitive features. Information on cognitive function is available for 61 individuals. Eight had normal intellect; 52 individuals had variable intellectual disability (ID), including the following:
- Mild ID (30/61). Children attend mainstream school but need some extra help – e.g., a statement of educational needs – and are expected to live independently as adults and likely to have their own family.
- Moderate ID (13/61). Children develop speech and need a high level of support in mainstream education but are likely to require special educational needs. While unlikely to live independently as adults, they may live in sheltered accommodation or with additional support.
- Severe ID (3/61). Individuals require special education during school and are likely to require considerable support during adulthood.
- Unclassified ID (6/61). Insufficient information was provided regarding degree of ID.
Behavioral issues including autistic features, phobias, and anxiety have been anecdotally reported [Tatton-Brown et al 2013].
Skeletal features
- Advanced bone age. Of 33 individuals evaluated, all had advanced bone age.
- Scoliosis was reported in 13 individuals and pectus abnormalities (excavatum or carinatum) in four individuals. Scoliosis ranged from severe (early-childhood onset requiring surgical intervention) to mild (requiring monitoring but no therapeutic intervention).
- Camptodactyly. Some affected individuals had camptodactyly of the fingers, some had camptodactyly of the toes, and some had camptodactyly of fingers and toes. On occasion, the toe camptodactyly required surgical correction.
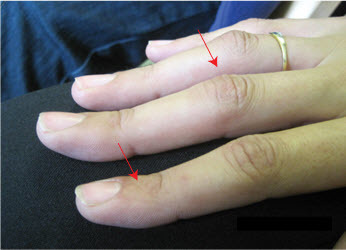
- Adult boutonniere deformity. Several adults developed hyperextension of the distal interphalangeal joints and flexion of the proximal interphalangeal joints of the hands analogous to a mild boutonniere deformity (see Figure 2).
- Talipes equinovarus. Eight individuals had talipes equinovarus ranging from fixed and bilateral (requiring surgery) in two individuals to mild (unilateral that resolved with physiotherapy) in three.
Figure 2.
Mild hyperextension of the distal interphalangeal joints and flexion of the proximal interphalangeal joints in a woman age 22 years with a heterozygous EZH2 pathogenic variant
Connective tissue features
- Ligamentous laxity. While ligamentous laxity with associated joint hypermobility and pes planus is common, it is not usually reported unless complicated by joint pain. Individuals with EZH2-related overgrowth are frequently reported to have poor coordination that may be (at least partially) attributable to lax ligaments.
- Skin that was soft and doughy to the touch has been reported in 23 of 43 affected children.
- Umbilical hernia has been seen in 28 of 57 children and was sufficiently large to require surgery in eight neonates.
Poor feeding has been reported in 10 of 28 neonates, including one who required nasogastric tube feeding for two weeks. Although poor feeding may be attributable to neonatal hypotonia, this was only reported in three of the infants with poor feeding.
Hoarse, low-pitched cry was reported in 16 of 35 affected infants.
Tumors have been reported in seven of 68 affected individuals [Tatton-Brown et al 2013, Usemann et al 2016, Cohen et al 2016, Oh et al 2023]. A summary of reported tumor types is provided in Table 3.
Table 3.
Tumor Types Reported in Individuals with EZH2-Related Overgrowth
Additional clinical features reported in a small number of individuals (and therefore possibly not associated with the EZH2 pathogenic variant) are included for completeness:
- Café au lait macules (2 individuals), hemangioma (4 individuals), pigmented nevi (3 individuals)
- Large hands and feet (1 individual)
- Hypermetropia (hyperopia; 3 individuals), strabismus (3 individuals), myopia (1 individual)
- Hydrocele (2 individuals), cryptorchidism (1 individual), hypospadias (1 individual)
- Cleft palate (3 individuals)
- Hearing loss (conductive and sensorineural; 3 individuals)
- Cardiac anomalies (4 individuals), including mitral valve prolapse (1 individual), ventricular septal defect (2 individuals), and patent ductus arteriosus (1 individual)
- Gastroesophageal reflux (1 individual), hiatal hernia (1 individual)
- Neonatal hypoglycemia (2 individuals)
- Neonatal hypocalcemia (2 individual)
Genotype-Phenotype Correlations
No genotype-phenotype correlations are evident among individuals reported with EZH2-related overgrowth, as findings along the entire phenotypic spectrum have been observed in individuals with heterozygous truncating or missense pathogenic variants in EZH2, within or outside the conserved SET domain (see Molecular Genetics).
Penetrance
Data are currently insufficient to determine the penetrance of EZH2 germline pathogenic variants. However, given the subtlety of the phenotype in some individuals with a pathogenic EZH2 variant, the penetrance for some EZH2 pathogenic variants may be reduced [Tatton-Brown et al 2013].
Nomenclature
Weaver syndrome is named after David Weaver, who reported two boys with accelerated osseous maturation, unusual facies, and camptodactyly [Weaver et al 1974].
Prevalence
As individuals with a mild phenotype may escape clinical diagnosis, it is currently difficult to estimate the prevalence of EZH2-related overgrowth.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in EZH2.
Sporadic tumors (including hematopoietic malignancies) occurring in the absence of any other findings of EZH2-related overgrowth frequently contain a somatic variant in EZH2 that is not present in the germline. In these circumstances predisposition to these tumors is not heritable.
Differential Diagnosis
Significant overlap in findings is observed between EZH2-related overgrowth, Sotos syndrome (associated with pathogenic variants in NSD1), Cohen-Gibson syndrome (associated with pathogenic variants in EED), and Imagawa-Matsumoto syndrome (associated with pathogenic variants in SUZ12).
Additional disorders of interest in the differential diagnosis of EZH2-related overgrowth are summarized in Table 4.
Table 4.
Disorders to Consider in the Differential Diagnosis of EZH2-Related Overgrowth
Management
No clinical practice guidelines for EZH2-related overgrowth have been published. In the absence of published guidelines, the following recommendations are based on the authors' personal experience managing individuals with this disorder.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with EZH2-related overgrowth, the evaluations summarized Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
EZH2-Related Overgrowth: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
There is no cure for EZH2-related overgrowth. Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 6). If additional clinical issues are detected through the history and/or examination, the appropriate specialist referral(s) should be made.
Table 6.
EZH2-Related Overgrowth: Treatment of Manifestations
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, Botox®, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Neurobehavioral/Psychiatric Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 7 are recommended.
Table 7.
EZH2-Related Overgrowth: Recommended Surveillance
Agents/Circumstances to Avoid
Histone-lysine N-methyltransferase EZH2 (EZH2), encoded by EZH2, is an enzymatic catalytic subunit of the polycomb repressive complex 2 (PRC2) [Duan et al 2020]. These proteins contribute to cell cycle regulation, apoptosis, and DNA damage repair and have therefore been identified as targets for cancer therapies [Duan et al 2020]. The efficacy and side effect profile of EZH2 inhibitors and PRC2 inhibitors may be altered in individuals with EZH2-related overgrowth (see EED-Related Overgrowth).
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
In general, pregnancies in which the mother and/or fetus has a heterozygous EZH2 pathogenic variant are uncomplicated. Families and their health care providers should be made aware that an affected baby may be large so that appropriate delivery plans can be made; in addition, information about the EZH2-related overgrowth phenotype should be provided.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
EZH2-related overgrowth is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Some individuals diagnosed with EZH2-related overgrowth have an affected parent.
- Some individuals diagnosed with EZH2-related overgrowth have the disorder as the result of a de novo EZH2 pathogenic variant. The proportion of individuals with EZH2-related overgrowth caused by a de novo pathogenic variant is unknown.
- Of 68 individuals with EZH2 pathogenic variants, 34 individuals represented simplex cases (i.e., the only family member known to be affected) and had a de novo pathogenic variant,18 individuals had a familial pathogenic variant, and 16 individuals could not be confirmed to have a de novo or a familial pathogenic variant as parental testing was not performed or clinical information was not available from a parent [Gibson et al 2012, Al-Salem et al 2013, Tatton-Brown et al 2013, Cohen et al 2016, Usemann et al 2016, Lui et al 2018, Griffiths et al 2019, Turkkahraman et al 2021].
- If the proband appears to be the only affected family member, molecular genetic testing is recommended for the parents of the proband to evaluate their genetic status and inform recurrence risk assessment.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
* A parent with somatic and germline mosaicism for an EZH2 pathogenic variant may be mildly/minimally affected. - The family history of some individuals diagnosed with EZH2-related overgrowth may appear to be negative because of failure to recognize the disorder in a parent with a milder phenotype. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is known to have the EZH2 pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. The phenotype in individuals who inherit a familial EZH2 pathogenic variant cannot be predicted.
- If the EZH2 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the EZH2 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband is unclear because of the possibility of reduced penetrance in a heterozygous parent or parental germline mosaicism.
Offspring of a proband. Each child of an individual with EZH2-related overgrowth has a 50% chance of inheriting the EZH2 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has an EZH2 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the EZH2 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal and preimplantation genetic testing. While most health care professionals would consider use of prenatal and preimplantation genetic testing to be a personal decision, discussion of these issues can be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- Child Growth FoundationUnited KingdomPhone: 0208 995 0257Email: nfo@childgrowthfoundation.org
- MAGIC FoundationPhone: 630-836-8200Email: contactus@magicfoundation.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
EZH2-Related Overgrowth: Genes and Databases
Table B.
OMIM Entries for EZH2-Related Overgrowth (View All in OMIM)
Molecular Pathogenesis
Histone-lysine N-methyltransferase EZH2 (EZH2) is a histone methyltransferase with a critical SET (su(var)3-9, enhancer of zeste, trithorax) domain, a pre-SET CXC domain, and two additional SANT (Sw13, Ada2, N-cor TFIIIB) domains [Wu et al 2013]. In combination with polycomb protein SUZ12 (SUZ12) and polycomb protein EED (EED), which form the polycomb repressor complex 2 (PCR2), EZH2 acts to repress transcription through the methylation of lysine residue 27 of histone 3, a function catalyzed by the SET domain [Cao et al 2002]. The protein alterations and the mechanism by which pathogenic EZH2 missense and truncating variants cause overgrowth are currently unknown. It is, however, noteworthy that pathogenic missense variants are the primary mutational mechanism; the few pathogenic truncating variants identified to date target the final exon and thus are likely to escape nonsense-mediated RNA decay. Current evidence suggests that EZH2-related overgrowth may be caused by impaired histone methyltransferase function [Cohen et al 2016, Lui et al 2018].
Mechanism of disease causation. Functional studies suggest that EZH2 pathogenic variants cause either partial or complete loss of protein function, resulting in impaired histone methyltransferase activity [Cohen et al 2016, Imagawa et al 2017, Lee et al 2018, Lui et al 2018]. However, alternative mechanisms of disease causation are theoretically possible [Cyrus et al 2019].
Epigenetic mutational signature analysis has shown that EZH2 gain-of-function variants may cause transcriptional changes that result in growth restriction, in contrast to EZH2-related overgrowth due to loss-of-function variants [Choufani et al 2020].
Further functional studies are needed to determine the precise mechanism of disease causation in EZH2-related overgrowth and other PRC2-related syndromes including Cohen-Gibson syndrome (see EED-Related Overgrowth) and Imagawa-Matsumoto syndrome.
EZH2-specific laboratory technical considerations. Based on a small number of cases, current data (in which the distribution of EZH2 variants in cases vs controls, conservation of the SET domain residues, and critical function of the SET domain in mediating histone methyltransferase activity were analyzed) suggest that SET domain missense variants are likely pathogenic.
Table 8.
EZH2 Pathogenic Variants Referenced in This GeneReview
Chapter Notes
Author History
Sharon Ocansey, MBBS, BSc, MSc (2024-present)
Nazneen Rahman, BM BCh, PhD; Institute of Cancer Research (2013-2024)
Katrina Tatton-Brown, BM BCh, MD (2013-present)
Revision History
- 21 March 2024 (gm) Comprehensive update posted live
- 2 August 2018 (bp) Comprehensive update posted live
- 6 August 2015 (me) Comprehensive update posted live
- 18 July 2013 (me) Review posted live
- 23 January 2013 (ktb) Original submission
References
Literature Cited
- Al-Salem A, Alshammari MJ, Hassan H, Alazami AM, Alkuraya FS. Weaver syndrome and defective cortical development: a rare association. Am J Med Genet A. 2013;161A:225–7. [PubMed: 23239504]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298:1039–43. [PubMed: 12351676]
- Choufani S, Gibson WT, Turinsky AL, Chung BH, Wang T, Garg K, Vitriolo A, Cohen AS, Cyrus S, Goodman S, Chater-Diehl E. DNA methylation signature for EZH2 functionally classifies sequence variants in three PRC2 complex genes. Am J Hum Genet. 2020;106:596-610. [PMC free article: PMC7212265] [PubMed: 32243864]
- Cohen AS, Yap DB, Lewis ME, Chijiwa C, Ramos-Arroyo MA, Tkachenko N, Milano V, Fradin M, McKinnon ML, Townsend KN, Xu J, Van Allen MI, Ross CJ, Dobyns WB, Weaver DD, Gibson WT. Weaver syndrome-associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Hum Mutat. 2016;37:301–7. [PMC free article: PMC4832389] [PubMed: 26694085]
- Cottereau E, Mortemousque I, Moizard MP, Bürglen L, Lacombe D, Gilbert-Dussardier B, Sigaudy S, Boute O, David A, Faivre L, Amiel J, Robertson R, Viana Ramos F, Bieth E, Odent S, Demeer B, Mathieu M, Gaillard D, Van Maldergem L, Baujat G, Maystadt I, Héron D, Verloes A, Philip N, Cormier-Daire V, Frouté MF, Pinson L, Blanchet P, Sarda P, Willems M, Jacquinet A, Ratbi I, Van Den Ende J, Lackmy-Port Lis M, Goldenberg A, Bonneau D, Rossignol S, Toutain A. Phenotypic spectrum of Simpson-Golabi-Behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet C Semin Med Genet. 2013;163C:92–105. [PubMed: 23606591]
- Cyrus S, Burkardt D, Weaver DD, Gibson WT. PRC2-complex related dysfunction in overgrowth syndromes: A review of EZH2, EED, and SUZ12 and their syndromic phenotypes. Am J Med Genet C Semin Med Genet. 2019;181:519-31. [PubMed: 31724824]
- Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M, Tomkins S, Hughes HE, Cole TR, Rahman N. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet. 2003;72:132–43. [PMC free article: PMC378618] [PubMed: 12464997]
- Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. 2020;13:104. [PMC free article: PMC7385862] [PubMed: 32723346]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, Marra MA., FORGE Canada Consortium. Chitayat D, Boycott KM, Weaver DD, Jones SJ. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet. 2012;90:110–8. [PMC free article: PMC3257956] [PubMed: 22177091]
- Griffiths S, Loveday C, Zachariou A, Behan LA, Chandler K, Cole T, D'Arrigo S, Dieckmann A, Foster A, Gibney J, Hunter M. EED and EZH2 constitutive variants: a study to expand the Cohen-Gibson syndrome phenotype and contrast it with Weaver syndrome. Am J Med Genet A. 2019;179:588-94. [PubMed: 30793471]
- Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, Ryo A, Sato Y, Sanefuji M, Ihara K, Takada Y, Nishimura G, Saitsu H, Mizuguchi T, Miyatake S, Nakashima M, Miyake N, Soejima H, Matsumoto N. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum Mutat. 2017;38:637–48. [PubMed: 28229514]
- Imagawa E, Seyama R, Aoi H, Uchiyama Y, Marcarini BG, Furquim I, Honjo RS, Bertola DR, Kim CA, Matsumoto N. Imagawa-Matsumoto syndrome: SUZ12-related overgrowth disorder. Clin Genet. 2023;103:383-91. [PubMed: 36645289]
- Klaassens M, Morrogh D, Rosser EM, Jaffer F, Vreeburg M, Bok LA, Segboer T, van Belzen M, Quinlivan RM, Kumar A, Hurst JA, Scott RH. Malan syndrome: Sotos-like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. Eur J Hum Genet. 2015;23:610–5. [PMC free article: PMC4402637] [PubMed: 25118028]
- Lee CH, Yu JR, Kumar S, Jin Y, LeRoy G, Bhanu N, Kaneko S, Garcia BA, Hamilton AD, Reinberg D. Allosteric activation dictates PRC2 activity independent of its recruitment to chromatin. Mol Cell. 2018;70:422-34.e6. [PMC free article: PMC5935545] [PubMed: 29681499]
- Lui JC, Barnes KM, Dong L, Yue S, Graber E, Rapaport R, Dauber A, Nilsson O, Baron J. Ezh2 mutations found in the Weaver overgrowth syndrome cause a partial loss of H3K27 histone methyltransferase activity. J Clin Endocrinol Metab. 2018;103:1470–8. [PMC free article: PMC6276576] [PubMed: 29244146]
- Malan V, Rajan D, Thomas S, Shaw AC, Louis Dit Picard H, Layet V, Till M, van Haeringen A, Mortier G, Nampoothiri S, Puseljic S, Legeai-Mallet L, Carter NP, Vekemans M, Munnich A, Hennekam RC, Colleaux L, Cormier-Daire V. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a Sotos-like or a Marshall-Smith syndrome. Am J Hum Genet. 2010;87:189–98. [PMC free article: PMC2917711] [PubMed: 20673863]
- Oh I, Kim B, Lee JS, Kim MJ, Im Cho S, Park SS, Seong MW. First Korean case of Weaver syndrome along with neuroblastoma and genetic confirmation of EZH2 variant. Lab Med Online. 2023;13:48-52.
- Priolo M, Schanze D, Tatton-Brown K, Mulder PA, Tenorio J, Kooblall K, Acero IH, Alkuraya FS, Arias P, Bernardini L, Bijlsma EK, Cole T, Coubes C, Dapia I, Davies S, Di Donato N, Elcioglu NH, Fahrner JA, Foster A, González NG, Huber I, Iascone M, Kaiser AS, Kamath A, Liebelt J, Lynch SA, Maas SM, Mammì C, Mathijssen IB, McKee S, Menke LA, Mirzaa GM, Montgomery T, Neubauer D, Neumann TE, Pintomalli L, Pisanti MA, Plomp AS, Price S, Salter C, Santos-Simarro F, Sarda P, Segovia M, Shaw-Smith C, Smithson S, Suri M, Valdez RM, Van Haeringen A, Van Hagen JM, Zollino M, Lapunzina P, Thakker RV, Zenker M, Hennekam RC. Further delineation of Malan syndrome. Hum Mutat. 2018;39:1226–37. [PMC free article: PMC6175110] [PubMed: 29897170]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR. UK10K Consortium, Hurles ME. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Schanze D, Neubauer D, Cormier-Daire V, Delrue MA, Dieux-Coeslier A, Hasegawa T, Holmberg EE, Koenig R, Krueger G, Schanze I, Seemanova E, Shaw AC, Vogt J, Volleth M, Reis A, Meinecke P, Hennekam RC, Zenker M. Deletions in the 3' part of the NFIX gene including a recurrent Alu-mediated deletion of exon 6 and 7 account for previously unexplained cases of Marshall-Smith syndrome. Hum Mutat. 2014;35:1092–100. [PubMed: 24924640]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Suri T, Dixit A. The phenotype of EZH2 haploinsufficiency-1.2-Mb deletion at 7q36.1 in a child with tall stature and intellectual disability. Am J Med Genet A. 2017;173:2731–5. [PubMed: 28696078]
- Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, Waggoner D, Turkmen S, Cormier-Daire V, Irrthum A, Rahman N. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet. 2005;77:193–204. [PMC free article: PMC1224542] [PubMed: 15942875]
- Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte Sdel V, Ramsay E, Snape K, Murray A, Perdeaux ER, Seal S, Loveday C, Banka S, Clericuzio C, Flinter F, Magee A, McConnell V, Patton M, Raith W, Rankin J, Splitt M, Strenger V, Taylor C, Wheeler P, Temple KI, Cole T, Douglas J, Rahman N. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget. 2011;2:1127–33. [PMC free article: PMC3282071] [PubMed: 22190405]
- Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, Bird LM, Clericuzio CL, Cormier-Daire V, Cushing T, Flinter F, Jacquemont ML, Joss S, Kinning E, Lynch SA, Magee A, McConnell V, Medeira A, Ozono K, Patton M, Rankin J, Shears D, Simon M, Splitt M, Strenger V, Stuurman K, Taylor C, Titheradge H, Van Maldergem L, Temple IK, Cole T, Seal S, Rahman N, et al. Weaver syndrome and EZH2 mutations: clarifying the clinical phenotype. Am J Med Genet A. 2013;161A:2972–80. [PubMed: 24214728]
- Turkkahraman D, Sakarya AN, Randa NC. A novel EZH2 gene variant in a case of Weaver syndrome with postaxial polydactyly. Am J Med Genet A. 2021;185:2234-7. [PubMed: 33788986]
- Usemann J, Ernst T, Schäfer V, Lehmberg K, Seeger K. EZH2 mutation in an adolescent with Weaver syndrome developing acute myeloid leukemia and secondary hemophagocytic lymphohistiocytosis. Am J Med Genet A. 2016;170A:1274–7. [PubMed: 26762561]
- Weaver DD, Graham CB, Thomas IT, Smith DW. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J Pediatr. 1974;84:547–52. [PubMed: 4366187]
- Wu H, Zeng H, Dong A, Li F, He H, Senisterra G, Seitova A, Duan S, Brown PJ, Vedadi M, Arrowsmith CH, Schapira M. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS One. 2013;8:e83737. [PMC free article: PMC3868588] [PubMed: 24367611]
Publication Details
Author Information and Affiliations
St George's University Hospital NHS Foundation Trust
London, United Kingdom
St George's University of London;
St George's University Hospital NHS Foundation Trust
London, United Kingdom
Publication History
Initial Posting: July 18, 2013; Last Update: March 21, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Ocansey S, Tatton-Brown K. EZH2-Related Overgrowth. 2013 Jul 18 [Updated 2024 Mar 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.