

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

The unicellular eukaryote Trypanosoma brucei (T. brucei) is the causative agent of human African trypanosomiasis (HAT), a disease that annually infects ~500,000 people in sub-Saharan Africa resulting in 50,000 – 70,000 deaths per year. Without treatment, HAT is fatal. Unfortunately, current treatments are limited in availability, have toxic side effects, are difficult to administer and are not well characterized in terms of their mechanism of action. Thus, the lack of affordable, safe, and effective therapies for those with African trypanosomiasis makes the identification of molecular target-specific chemotypes a priority in our effort to understand the mechanisms involved with parasite growth and viability, as well as for future therapeutic development. The probe identified from this effort, ML205, is a stable, small molecule possessing submicromolar activity (IC50 = 0.98 μM) against a defined T. brucei hexokinase 1 (rTbHK1) target. The probe was not toxic to mammalian cells (IMR-90 cells, EC50 > 25 μM) and mechanistic studies revealed that the probe operates with mixed inhibition with respect to ATP.
Assigned Assay Grant #: 1 R03 MH082340-01A1
Screening Center & PI: Pittsburgh Screening Center (MLSCN), John Lazo
Chemistry Center & PI: University of Kansas Specialized Chemistry Center, Jeffrey Aubé
Assay Submitter & Institution: James C. Morris, Clemson University
PubChem Summary Bioassay Identifier (AID): AID 2600
Probe Structure & Characteristics
| CID/ML# | Target Name | IC50/EC50 (nM) [SID, AID] | Anti-target Name(s) | IC50/EC50 (μM) [SID, AID] | Fold Selective | Secondary Assay(s) Name: IC50/EC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 46931017 ML205 | TbHK1 | IC50 = 976 nM (SID 99437306, AID 2230) | G6PDH and hGlk | G6PDH IC50 > 25000 nM; (SID 99437306, AID 2579) hGlk IC50 =48300 nM; (SID 99437306, AID 492951) | G6PDH:TbHK1 = 25.5 hGlk:TbHK1 = 49.3 | IMR-90 cell viability assay; EC50 > 25000 nM; (SID 99437306, AID 449725) |
Recommendations for scientific use of the probe
What limitations in current state of the art is the probe addressing: The unicellular eukaryote Trypanosoma brucei (T. brucei) is the causative agent of human African trypanosomiasis (HAT), a disease that annually infects ~500,000 people in sub-Saharan Africa resulting in 50,000 – 70,000 deaths per year. Disease manifestation consists of the haemolymphatic and neurological phases, the latter of which begins when the T. brucei parasites cross the blood brain barrier and invade the central nervous system. Without treatment, HAT is fatal. Unfortunately, current treatments are limited in availability, have toxic side effects, are difficult to administer and are not well characterized in terms of their mechanism of action. There are only four drugs approved for treatment of HAT. Suramin and pentamidine, however, are not effective against the neurological stage of HAT while melarsoprol leads to fatal complications in 5–10% of patients receiving the drug. The most recently developed drug, eflornithine, is expensive and only efficacious against T. b. gambiense, but is curative in both stages of the disease. However, delivery of eflornithine is difficult, as the compound must be administered intravenously four times a day for 14 days. Thus, the lack of affordable, safe, and effective therapies for those with African trypanosomiasis makes the identification of molecular target-specific chemotypes a priority in our effort to understand the mechanisms involved with parasite growth and viability, as well as for future therapeutic development [1, 2]. The probe identified from this effort, ML205, is a stable, small molecule possessing submicromolar activity against a defined T. brucei hexokinase 1 (TbHK1) target with mixed inhibition with respect to ATP.
What will the probe be used for: The assay provider is currently using the probe to investigate the nature of TbHK1 inhibition and is interested to use this as the starting point for further characterization, aimed ultimately at demonstrating in vivo efficacy and providing a lead for advanced development in the treatment of HAT. The probe was the subject of a probe enhancement proposal, submitted to the NIH on October 13, 2010, and recently funded as a grant proposal. In this regard, the probe will be used as a means of demonstrating (a) TbHK1 on target activity via an orthogonal ATP-regeneration coupled assay, (b) impact on parasitic cell growth potential in a live/dead BSF assay, (c) efficacy against a related parasite using a whole parasite-based, Leishmania promastigote assay, (d) action on the in vivo target, (e) TbHK1 activity from T. brucei parasite lysate with IC50 similar to recombinant protein, (f) adequate in vivo PKDM properties leading to (g) showing efficacy in an acute infection mouse model. As outlined above, to truly impact this disease and the people affected by it, there is a dire need to identify better performing leads, further understand the mechanism by which compounds can inhibit T. brucei effectively, and ultimately halt the associated disease that is treated inadequately with currently available agents. This probe will establish a target-based approach of disease interference for HAT.
Who in the research community will use the probe: The assay provider will be using the probe for the purposes described above. The probe will be useful to groups interested in identifying resistance pathways in the parasite, particularly in collaboration with the powerful forward genetic tools available in the T. brucei system. Moreover, if the compound has efficacy in a mouse model, suggesting it is a lead compound that merits further development for this important neglected tropical-disease, interest will be wide-spread.
What is the relevant biology to which the probe can be applied: The development of lead compounds targeting various pharmacology for the treatment of human African trypanosomiasis was recently reviewed [3]. A TbHK1 small molecule probe is of particular interest because of the importance of glucose metabolism to the infectious bloodstream form (BSF) lifecycle stage of the parasite. Specifically, in the mammalian host, the T. brucei BSF parasite generates ATP exclusively through glycolysis. Hexokinase (HK), the first enzyme in glycolysis, catalyzes the transfer of the γ-phosphoryl group of ATP to glucose yielding glucose-6-phosphate. Several lines of evidence indicate that inhibition of TbHK1 activity in BSF parasites is lethal [4, 5]. Previously, the Assay Provider (Dr. James Morris, Clemson University) demonstrated that recombinant TbHK1 (rTbHK1) is inhibited by lonidamine (IC50 = 850 μM), an anti-cancer drug that targets tumor hexokinases, and the compound is toxic to T. brucei [6]. Furthermore, the HTS effort sponsored by this project has revealed potent anti-parasitics against the recombinant enzyme, suggesting that through careful SAR development, inhibitors of TbHK1 can be identified that will be useful lead compounds for the development of anti-trypanosomal agents.
1. Introduction
Probe Project Purpose: There is a significant need to identify new agents that effectively address human African trypanosomiasis (HAT), as current treatments are limited in number and availability, have toxic side effects, are difficult to administer or are not well characterized in terms of their mechanism of action. The purpose of this probe project was to identify a small molecule, single-digit micromolar range inhibitor of recombinant T. brucei hexokinase 1 (TbHK1) which would provide a good lead for further development and establish a target-based approach of potentially treating HAT.
Prior Art Status: There are only four drugs approved for treatment of HAT (Fig. 1). However, suramin and pentamidine are not effective against the neurological stage of HAT and melarsoprol leads to fatal complications in 5–10% of patients receiving the drug. The most recently developed drug, eflornithine, is expensive and only efficacious against T. b. gambiense (one of three known species), but is curative in both stages of the disease. However, delivery of eflornithine is difficult, as the compound must be administered intravenously four times a day for 14 days. Thus, the lack of affordable, safe, and effective therapies for those with African trypanosomiasis makes the identification of molecular target-specific chemotypes a priority in our effort to understand the mechanisms involved with parasite growth and viability, as well as for future therapeutic development [1, 2].
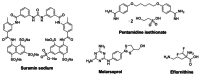
Figure 1
Currently available marketed drugs for treatment of human African trypanosomiasis.
Recently, the Assay Provider (Dr. James Morris, Clemson University) demonstrated that recombinant TbHK1 (rTbHK1) is inhibited by lonidamine (IC50 = 850 μM), an anti-cancer drug that targets tumor hexokinases (Fig. 2) [6]. Lonidamine also inhibits HK activity from whole parasite lysate (IC50 = 965 μM) and is toxic to cultured bloodstream form parasites (LD50 = 50 μM). Myristic acid and quercetin were also reported to inhibit rTbHK1 at IC50 values of 78 μM and 85 μM, respectively [7]. Thus, for the duration of this project, to our knowledge no potent and specific small molecule inhibitors of TbHK1 existed.

Figure 2
Known TbHK1 inhibitors and preliminary associated data.
The Assay Provider recently undertook an independent study of quercetin and determined the rTbHK1 IC50 to be 4.1 ± 0.8 μM [8]. Given the discrepancy of rTbHK1 inhibition data associated with quercetin, the KU SCC obtained a commercial source of the material, purified it by reverse-phase chromatography, and performed a rigorous analytical analysis for structural integrity and purity assessment. This sample was then evaluated in the same rTbHK1 in vitro assay that was used for assessment of analogs for this project. Additionally, the Pittsburgh Screening Center supplied its own sample of quercetin in a side-by-side analysis. Depending on the source of the material, there was an observed variance in rTbHK1 inhibition (Table 1).
Table 1
Comparison of data obtained with variable quercetin sources.
While the team has not definitely established the underlying cause for the observed range in activity, it has been reasoned that the well-established antioxidant nature of the flavinol structure renders this class of compounds susceptible to gradual transformation which could give rise to contaminants in the sample. A rigorous purification and analysis of material prior to assay revealed an rTbHK1 IC50 = 22.4 μM. The team is interested in following up further on suspected contaminants in non-purified samples that might be contributing to the observed low micromolar inhibition of TbHK1. In any event, quercetin is the most relevant TbHK1 inhibitor due to its activity to which a new probe should be compared. Like lonidamine, quercetin (unpurified) has also been shown to inhibit hexokinase activity from whole parasite lysate (IC50 = 24 μM), and is toxic to cultured bloodstream form parasites (LD50 = 7.5 μM) [8]. Importantly, quercetin has been reported to engage other mammalian enzymes such as a tyrosine protein kinase, a phosphorylase kinase [9] a phosphatidyl 3-kinase [10] and a DNA topoisomerase [11]. The inhibition of these targets is in the IC50 range of 3–300 μM, which coincides with that for TbHK1, thereby suggesting that there may be only limited opportunities to increase the therapeutic range for the compound [8]. Moreover, a more selective probe with few off-target effects would greatly facilitate the study of how to effectively inhibit TbHK1 and provide a better lead positioned for development of HAT therapeutics.
2. Materials and Methods
2.1. Assays
A. Primary HTS TbHK1 Assay
Descriptive Name: Inhibition of TbHK1 using glucose 6-phosphate dehydrogenase and monitoring depletion of NAD+ at a single concentration (10 μM).
Purpose: The purpose of this assay is to identify compounds that act as TbHK1 small molecule inhibitors in HTS format at a single concentration of 10 μM.
Summary AID: AID 2600
Assigned AID: AID 1430
Screening Responsibility: Pittsburgh Molecular Libraries Screening Center (MLSCN), John S. Lazo
Non-phenotypic Assay; Non-multiplexed Assay; BSL: BSL1
Assay Format: biochemical
Assay Type: enzymatic
Assay Method: endpoint assay
Assay Detection: absorbance
Target type: kinase
Assay Description: The biochemical, absorbance-based assay used for HTS implementation employs recombinant TbHK1 protein and glucose and ATP as the substrates in a coupled reaction. In this assay, commercially available glucose 6-phosphate dehydrogenase is included as a coupling enzyme to reduce NAD+ to NADH (which can be monitored spectrophotometrically at 340 nm) during the oxidation of glucose-6-P to 6-phosphogluconic acid. This assay was performed in a 384 well format and each assay plate contained 32 Maximum (vehicle; 1% DMSO), 24 Minimum (133 μM) and 8 IC50 control (1.3 μM) wells. These controls were used to derive the Z-factor and signal to background of each assay plate. Assay plates were passed and/failed according to the derived Z-factors. To account for possible inhibition of the other enzymes in the primary coupled reaction, putative inhibitors were screened to assess their impact against glucose-6-phosphate dehydrogenase counter-screening assay [7].
B. Primary TbHK1 Assay for SAR
Descriptive Name: Inhibition of TbHK1 using glucose 6-phosphate dehydrogenase and monitoring depletion of NAD+- 10 point concentration response assay.
Purpose: This assay is the same as the HTS screen; however, the compounds are evaluated over 10–20 concentrations in a dose response format.
Summary AID: AID 2600
Assigned AID: AID 1632
Screening Responsibility: Pittsburgh Molecular Libraries Screening Center (MLSCN), John S. Lazo
Non-phenotypic Assay; Non-multiplexed Assay; BSL: BSL1
Assay Format: biochemical
Assay Type: enzymatic
Assay Method: endpoint assay
Assay Detection: absorbance
Target Type: kinase
Assay Description: The biochemical, absorbance-based assay used for HTS implementation employs recombinant TbHK1 protein and glucose and ATP as the substrates in a coupled reaction. In this assay, commercially available glucose 6-phosphate dehydrogenase is included as a coupling enzyme to reduce NAD+ to NADH (which can be monitored spectrophotometrically at 340 nm) during the oxidation of glucose-6-P to 6-phosphogluconic acid. This assay was performed in a 384 well format and each assay plate contained 32 Maximum (vehicle; 1% DMSO), 24 Minimum (133 μM) and 8 IC50 control (1.3 μM) wells. These controls were used to derive the Z-factor and signal to background of each assay plate. Assay plates were passed and/failed according to the derived Z-factors. To account for possible inhibition of the other enzymes in the primary coupled reaction, putative inhibitors were screened to assess their impact against glucose-6-phosphate dehydrogenase counter-screening assay. Compounds are evaluated at 10–20 concentrations [7].
C. Secondary G6PDH Assay
Descriptive Name: G6PDH Coupled Enzyme Counter Screen
Purpose: The purpose of this assay is to identify and remove false positives from consideration that interfere with the primary assay by interfering with the reporter enzyme, G6PDH.
Summary AID: AID 2600
Assigned AID: AID 2516
Screening Responsibility: Pittsburgh Molecular Libraries Screening Center (MLSCN), John S. Lazo
Non-phenotypic Assay; Non-multiplexed Assay; BSL: BSL1
Assay Format: biochemical
Assay Type: enzymatic
Assay Method: endpoint assay
Assay Detection: absorbance
Target type: cellular pathway
Assay Description: This assay is a modification of the primary assay, with glc6P supplemented in the reaction and ATP, glucose and TbHK1 removed in order to measure the activity of the reporter enzyme, G6PDH. For primary screening, compounds will be excluded if inhibit G6DPH >50% of signal readout. For IC50 determinations, G6DPH inhibition would be 10x greater than inhibition of TbHK1 [7].
D. Secondary IMR-90 Assay
Descriptive Name: Human IMR90 Growth Inhibition Assay
Purpose: The purpose of this assay is to assess impact of compounds on human cell line while showing efficacious toxicity against T. brucei.
Summary AID: AID 2600
Assigned AID: AID 449725
Screening Responsibility: Pittsburgh Molecular Libraries Screening Center (MLSCN), John S. Lazo
Phenotypic Assay; Non-multiplexed Assay; BSL: BSL1
Assay Format: live, cell-based
Assay Type: viability/toxicity
Assay Method: endpoint assay
Assay Detection: fluorescence intensity
Target type: cellular pathway
Assay Description: This is a Alamar-blue based viability assay. IMR90 cells are cultured and maintained according to ATCC specifications (ATCC, Manassas, VA). IMR90 line drug susceptibility assays are performed in final volumes of 25 μL using a 384-well assay format. Briefly, 1,000 cells/22 μL in complete culture medium are seeded into each well of 384-well microtiter plates using a Titertek MAPC-2 bulk dispenser. Test and control compounds are added to individual wells (3 μL/well). Vehicle and positive controls are 1% DMSO and 10% DMSO, respectively (final well concentrations). Assay plates are incubated for 44h at 37°C in the presence of 5% CO2 and growth inhibitory effects are determined after the addition of 5 μL/well of Alamar blue reagent (incubate 4 hr). Data are captured on a Molecular Devices SpectraMax M5 platereader [7].
E. Secondary BSF live/dead Assay - NOT REQUIRED FOR PROBE CRITERIA
Descriptive Name: T. brucei parasite toxicity assay
Purpose: The purpose of this assay is to assess compounds in their ability to inhibit cell growth of T. brucei parasites.
Screening Responsibility: Assay provider, James Morris, Clemson University
Phenotypic Assay; Non-multiplexed Assay; BSL: BSL2
Assay Format: live, cell-based
Assay Type: viability/toxicity
Assay Method: endpoint assay
Assay Detection: fluorescence intensity
Target type: cellular pathway
Assay Description: This assay is an adaptation to a 96-well format of an Alamar blue-based cell viability assay (using Cell-Titer Blue) that measures the reduction of resazurin to resorufin by cellular processes. To determine the impact of TbHK1 inhibitors on cell growth, parasites are seeded at 5 × 103 BSF parasites (cell line 90–13, a 427 strain) into 96-well clear-bottomed polystyrene plates in 200 μl HMI-9 supplemented with 10% fetal bovine serum and 10% Serum Plus (Sigma-Aldrich, St. Louis, MO) and grown in the presence of compound (2 μl) or equivalently diluted carrier for 3 days in 5% CO2 at 37°C. CellTiter Blue (Promega, Madison WI) is added (20 μl) and the plates incubated an additional 3 hr under standard culture conditions. Fluorescence emission at 585 nm is then measured after excitation at 546 nm in a GENios microtiter plate reader (Phenix Research Products, Hayward CA) [7].
F. Secondary hGlk Assay
Descriptive Name: Human Glucokinase (hGlk) Counter Screen Assay
Purpose: The purpose of this assay is to identify compounds that are nonselective or possess potentially undesirable cross reactivity, due to inhibition of the related enzyme, hGlk.
Screening Responsibility: Assay provider, James Morris
Summary AID: AID 2600
Assigned AID: AID 492951
Non-phenotypic Assay; Non-multiplexed Assay; BSL: BSL1
Assay Format: biochemical
Assay Type: enzymatic
Assay Method: kinetic
Assay Detection: absorbance
Target type: kinase
Assay Description: This assay uses human HK 4 (i.e., human glucokinase, hGlk, GenBank accession no. BC001890) expressed from a cloned cDNA (OPEN Biosystems, Huntsville, AL) in pQE30. After sequencing, the plasmid is transformed into E. coli M15 (pREP) and cultures are grown to an OD600 of 0.9 in terrific broth and protein expression induced (3 hr, 37°C) with 1 mM IPTG, followed by purification by nickel affinity chromatography. Ideally, IC50s for hGlk will be >10 μM. Please note that while we prefer selective inhibitors of TbHK1, inhibition of the human enzyme does not indicate removal from consideration, as the trypanosome is exclusively reliant on TbHK1 for metabolism, while host organisms (including humans) have a varied metabolism and redundancies that may allow them to tolerate inhibition of related enzymes [7].
2.2. Probe Chemical Characterization
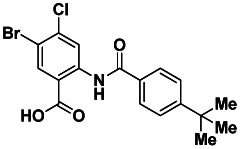
A. Probe Chemical Structure, Physical Parameters and Probe Properties

SID 99437306
CID 46931017
ML205
Molecular Formula: C18H17BrClNO3
Molecular Weight: 410.69 [g/mol]
Exact Mass: 409.01 [g/mol]
CLogP: 6.74
Topological Polar Surface Area: 66
Solubility (PBS buffer, pH 7.4): 25.6 μg/mL
Stability (% remaining after 48 h): 100%
Purity (LCMS, 215 nm): 100%
Melting Point: 273–276 °C
Physical State: white solid
B. Structure Verification and Purity: 1H NMR, 13C NMR, LCMS and HRMS
Proton and carbon NMR data for ML205 SID 99437306, CID 46931017: Detailed analytical methods and instrumentation are described in section 2.3, entitled “Probe Preparation” under general experimental and analytical details. The numerical experimental proton and carbon data are represented below, along with the associated spectra.
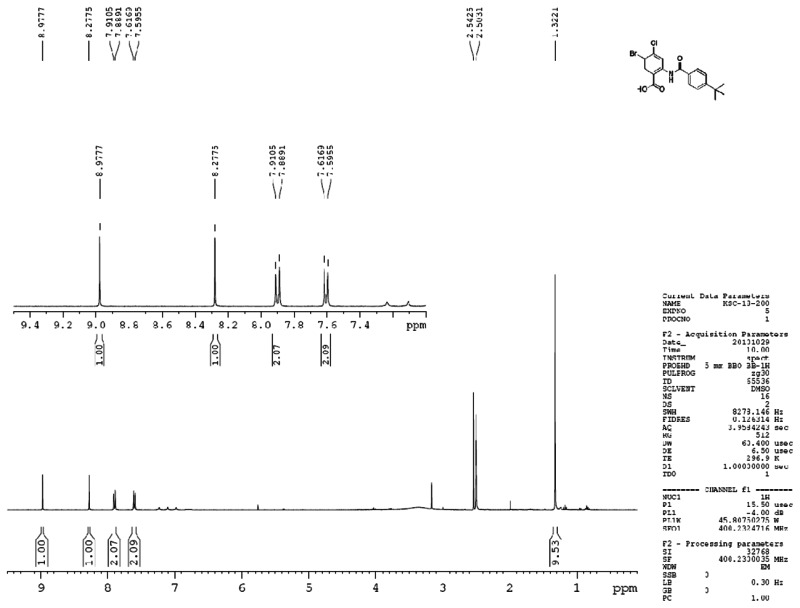
Figure 3Proton spectra for ML205, SID 99437306, CID 46931017
ML205, SID 99437306, CID 46931017 PROTON NMR DATA: 1H NMR (400 MHz, DMSO-d6) δ 13.27 (s, 1H), 8.98 (s, 1H), 8.28 (s, 1H), 7.90 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 1.32 (s, 9H).
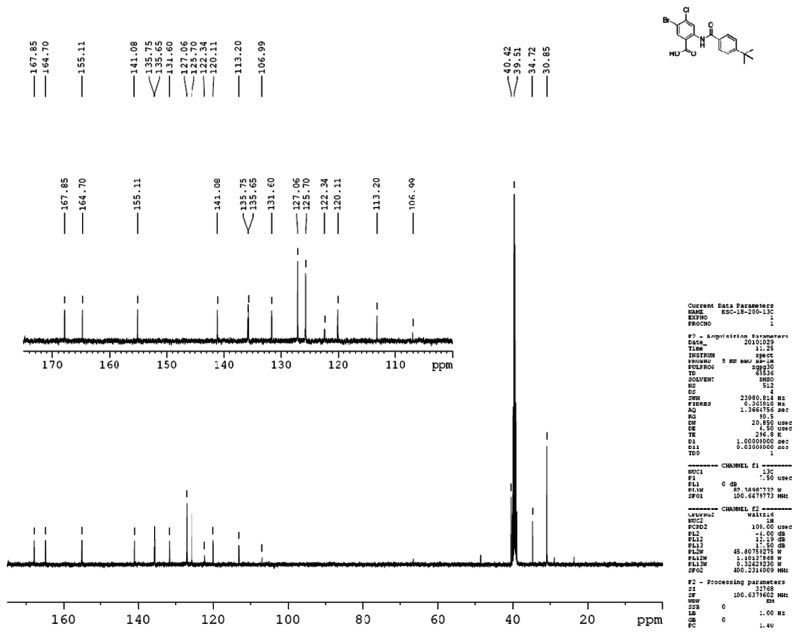
Figure 4Carbon spectra for ML205, SID 99437306, CID 46931017
ML205, SID 99437306, CID 46931017 CARBON NMR DATA: 13C NMR (100 MHz, DMSO-d6) δ 167.9, 164.7, 155.1, 141.1, 135.8, 135.7, 131.6, 127.1, 125.7, 122.3, 120.0, 113.2, 34.7, 30.9.
LCMS and HRMS data for ML205, SID 99437306, CID 46931017: Detailed analytical methods and instrumentation are described in section 2.3, entitled “Probe Preparation” under general experimental and analytical details. The numerical experimental LCMS and HRMS data are represented below, along with the associated spectra.
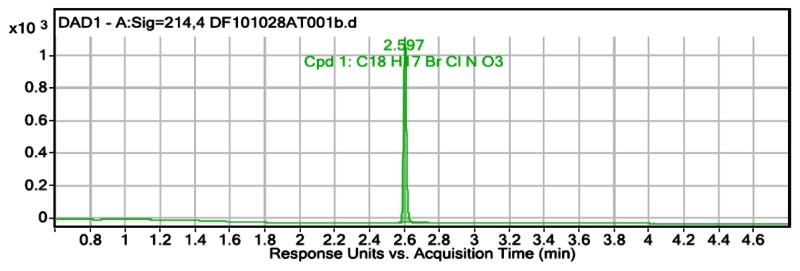
Figure 5LCMS purity data at 215 nm for ML205, SID 99437306, CID 46931017; LCMS retention time: 2.597 min; purity at 215 nm = 100%
User Chromatogram Peak List
| Peak # | Compound Name | RT | Height | Height % | Area | Area % | Area Sum % | Width |
|---|---|---|---|---|---|---|---|---|
| 1 | Cpd 1: C18 H17 Br Cl N O3 | 2.597 | 1119.43 | 100 | 1266.12 | 100 | 100 | 0.017 |
![Figure 6. HRMS data for ML205, SID 99437306, CID 46931017; HRMS m/z calculated for C18H17BrClNO3 [M+ - 1] 410.69, found 409.9995.](/books/NBK63599/bin/ml205f6.jpg)
Figure 6HRMS data for ML205, SID 99437306, CID 46931017; HRMS m/z calculated for C18H17BrClNO3 [M+ - 1] 410.69, found 409.9995
| Compound Label | RT | Mass | Abund | Formula | Tot Mass | Purity Value |
|---|---|---|---|---|---|---|
| Cpd 1: C18 H17 Br Cl N 03 | 2.592 | 409.0089 | 191057 | C18 H17 Br Cl N O3 | 409.008 | 100 |
C. Solubility
Aqueous solubility was measured in phosphate buffered saline (PBS) at room temperature (23°C). PBS by definition is 137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, 2 mM potassium phosphate monobasic and a pH of 7.4. The solubility of probe ML205, SID 99437306, CID 46931017 was determined to be 25.6 μg/mL [12].
D. Stability
Stability was measured at room temperature (23ºC) in PBS (no antioxidants or other protectants and DMSO concentration below 0.1%). The stability of probe compound ML205, SID 99437306, CID 46931017, determined as the percent of compound remaining after 48 hours, was 100% [12].
E. Synthetic Route
The probe and analogs were generally synthesized by the method shown (Fig. 8). Commercial acid chlorides were used when available. Otherwise, substituted benzoic acids 1 were converted to the corresponding acid chlorides 2 using oxalyl chloride. The crude acid chlorides were then treated with substituted aminobenzoates 3 in the presence of pyridine to afford amidobenzoates 4. Hydrolysis of the carboxylic esters provided the desired acids 5. Specific experimental details for the probe are detailed in section 2.3 below, entitled, “Probe Preparation.”

Figure 8
General synthetic route for probe and analogue synthesis. Reagents: (a) (COCl)2, DMF, DCM, 0 °C to rt, 2h; (b) pyridine, THF, 50 °C, 18 h; (c) LiOH, THF/H2O, rt, 3h.
If a requisite substituted benzoic acid or acid chloride was unavailable, the desired intermediates were prepared by Molander-type coupling of a halogenated benzoate or by Friedel-Crafts alkylation of the appropriate benzoate [13, 14].
F. Submission of Five Related Analogues to the MLSMR
Five analogues have been fully characterized and prepared in sufficient quantity for submission to the MLSMR. The five selected analogs and associated data are shown in Figure 9.

Figure 9
Five analogues selected for submission to support probe ML205, SID 99437306, CID 46931017 and associated data.
2.3. Probe Preparation
General experimental and analytical details: 1H and 13C NMR spectra were recorded on a Bruker AM 400 spectrometer (operating at 400 and 101 MHz respectively) in CDCl3 with 0.03% TMS as an internal standard or DMSO-d6. The chemical shifts (δ) reported are given in parts per million (ppm) and the coupling constants (J) are in Hertz (Hz). The spin multiplicities are reported as s = singlet, br. s = broad singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet and m = multiplet. The LCMS analysis was performed on an Agilent 1200 RRL chromatograph with photodiode array UV detection and an Agilent 6224 TOF mass spectrometer. The chromatographic method utilized the following parameters: a Waters Acquity BEH C-18 2.1 × 50mm, 1.7 μm column; UV detection wavelength = 214 nm; flow rate = 0.4ml/min; gradient = 5 – 100% acetonitrile over 3 minutes with a hold of 0.8 minutes at 100% acetonitrile; the aqueous mobile phase contained 0.15% ammonium hydroxide (v/v). The mass spectrometer utilized the following parameters: an Agilent multimode source which simultaneously acquires ESI+/APCI+; a reference mass solution consisting of purine and hexakis(1H, 1H, 3H-tetrafluoropropoxy) phosphazine; and a make-up solvent of 90:10:0.1 MeOH:Water:Formic Acid which was introduced to the LC flow prior to the source to assist ionization. The melting point was determined on a Stanford Research Systems OptiMelt apparatus.
Probe compound Ml205, SID 99437306, CID 46931017 was prepared by coupling 2-amino-5-bromo-4-chlorobenzoate with 4-tert-butylbenzoyl chloride, followed by hydrolysis (Figure 10).
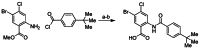
Figure 10
Assembly of probe compound ML205, SID 99437306, CID 46931017. Reagents: a) pyridine, THF, 50 °C, 18 h, 68%; b) LiOH, THF/H2O, rt, 3 h, 93%.
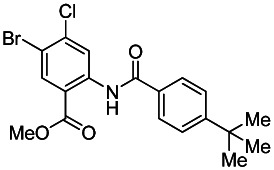
Methyl 2-(4-tert-butylbenzamido)-5-bromo-4-chlorobenzoate. Methyl 2-amino-5-bromo-4-chlorobenzoate (Ace Synthesis, CAS # 765211-09-4) and 4-tert-butylbenzoyl chloride (Sigma-Aldrich, CAS # 1710-98-1) were obtained commercially. In a 40 mL vial was combined sequentially methyl 2-amino-5-bromo-4-chlorobenzoate (0.21 g, 0.81 mmol, 1 eq.), 4-tert-butylbenzoyl chloride (0.22 mL, 1.21 mmol, 1.5 eq.) and pyridine (0.098 mL, 1.21 mmol, 1.5 eq.) in THF (2 mL). The resulting colorless solution was stirred under nitrogen at 50 °C for 18 hours. The reaction was removed from heat, cooled to rt, diluted with EtOAc (5 mL) and quenched with saturated aqueous NaHCO3 (5 mL). After extracting with EtOAc (2 × 5 mL), the crude material was adsorbed onto silica. Purification by silica gel chromatography (0–25% EtOAc:Hex ramp over 20 min) afforded the desired product methyl 2-(4-tert-butylbenzamido)-5-bromo-4-chlorobenzoate as a white solid (0.26 g, 0.55 mmol, 68% yield). 1H NMR (400 MHz, CDCl3) δ11.95 (s, 1H), 9.22 (s, 1H), 8.31 (s, 1H), 7.96 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.6 Hz, 2H), 3.99 (s, 3H), 1.36 (s, 9H).

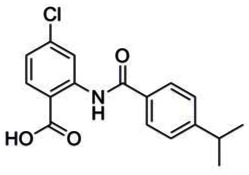
PROBE: 2-(4-tert-butylbenzamido)-5-bromo-4-chlorobenzoic acid. (ML205, SID 99437306, CID 46931017): To a mixture of methyl 2-(4-tert-butylbenzamido)-5-bromo-4-chlorobenzoate (0.26 g, 0.55 mmol, 1 eq.) and LiOH (0.092 g, 3.84 mmol, 7 eq.) in water (2 mL) was added THF (2 mL) to give a colorless solution. After stirring the solution for 3 h at rt, the reaction was acidified with 1.0 M aq. HCl to pH 2 – 3 and then extracted with CH2Cl2 (2 × 5 mL). The separated organic extracts were combined, dried (MgSO4), filtered and concentrated to produce the desired 2-(4-tert-butylbenzamido)-5-bromo-4-chlorobenzoic acid as a white solid (0.23 g, 0.57 mmol, 93 % yield). 1H NMR (400 MHz, DMSO-d6) δ 13.27 (s, 1H), 8.98 (s, 1H), 8.28 (s, 1H), 7.90 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 1.32 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ 167.9, 164.7, 155.1, 141.1, 135.8, 135.7, 131.6, 127.1, 125.7, 122.3, 120.0, 113.2, 34.7, 30.9. LCMS retention time: 2.592 min, purity at 215 nm = 100%. HRMS m/z calculated for C18H17BrClNO3 [M+ - 1] 410.69, found 409.99. White solid, mp 273 – 276 °C.
ANALOGUE 1: 4-chloro-2-(4-isopropylbenzamido)benzoic acid (SID 99222690/CID 45436131): Isolated 42 mg, yield 97% of 4-chloro-2-(4-isopropylbenzamido)benzoic acid as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 8.84 (d, J = 2.1 Hz, 1H), 8.06 (d, J = 8.6 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 8.3 Hz, 2H), 7.27 (dd, J1 = 8.6 Hz, J2 = 2.2 Hz, 1H), 2.99 (m, J = 6.9 Hz, 1H), 1.24 (d, J = 7.0 Hz, 6H). 13C NMR (125 MHz, DMSO-d6) δ169.37, 164.89, 153.33, 142.28, 138.73, 133.00, 131.66, 127.24, 127.04, 122.72, 119.06, 115.03, 33.43, 23.55. LCMS retention time: 2.327 min, purity at 215 nm = 98.2%. HRMS m/z calculated for C17H16ClNO3 [M+ + 1] 317.0818, found 318.0891. White solid, mp 217 – 220 °C.

ANALOGUE 2: 5-bromo-4-chloro-2-(4-methoxybenzamido)benzoic acid (SID 99380766/CID 46916165): Isolated 132 mg, yield 98 % of 5-bromo-4-chloro-2-(4-methoxybenzamido)benzoic acid as a white solid. 1H NMR (400 MHz, DMSO-d6) δ12.10 (s, 1H), 8.98 (s, 1H), 8.26 (s, 1H), 7.90 (d, J = 8.8 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 168.22, 164.38, 162.63, 141.22, 138.48, 135.48, 129.08, 125.82, 120.81, 117.02, 114.35, 113.77, 55.56. LCMS retention time: 2.345 min; purity at 215 nm = 100%. HRMS m/z calculated for C18H18ClNO3 [M+ + 1] 382.9565, found 383.9633. White solid, mp 258 – 261 °C.

ANALOGUE 3: 2-(4-tert-butylbenzamido)-4,5-dichlorobenzoic acid. (SID 99437306/CID 46947875 was prepared according to the following scheme:

Reagents: a) NaOH, NaOCl, MeOH, 80 °C, 2 h, 60%; b) 4-tert-butylbenzoyl chloride, TMS-diazomethane, CH2Cl2/MeOH, rt 30 min, 87%; c) CH3CN, 150 °C, 30 min, MW, 32%; d) LiOH, THF/H2O, rt, 3 h, 96%.

2-Amino-4,5-dichlorobenzoic acid. The starting material 4,5-dichlorophthalimide (CAS# 15997-89-4) was purchased from Sigma-Aldrich. In a vial was added the 4,5-dichlorophthalimide (1.93 g, 8.92 mmol, 1 eq.) and MeOH (10 mL). The 10.0 M NaOH (3.6 mL, 35.6 mmol, 4 eq.) and 10 % w/w NaOCl solution (6.08 mL, 9.81 mmol, 1.1 eq) were added and the reaction stirred for 2 hours at 80 °C. The reaction was then cooled to rt and was poured into 220 mL of 1.0 M HCl. After extracting with CHCl3 (3 × 200 mL) the crude material was adsorbed onto silica. Purification by silica gel chromatography (0–100% EtOAc:Hex ramp over 20 min) afforded the desired product 2-amino-4,5-dichlorobenzoic acid as a white solid (1.09 g, 5.30 mmol, 60% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.77 (s, 1H), 7.10 (s, 1H).
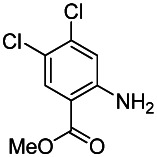
Methyl 2-amino-4,5-dichlorebenzoate. To a vial was added 2-amino-4,5-dichlorobenzoic acid (0.66 g, 3.21 mmol, 1 eq.) and DCM/MeOH (3 mL/2 mL). A solution of 2.0 M TMS-diazomethane in hexanes (2.89 mL, 5.78 mmol, 1.8 eq.) was added dropwise and the reaction stirred for 30 minutes at rt. The crude material was then adsorbed onto silica. Purification by silica gel chromatography (0–20% EtOAc:Hex ramp over 20 min) afforded the desired product methyl 2-amino-4,5-dichlorobenzoate as a white solid (0.61 g, 2.80 mmol, 87% yield). 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H), 7.79 (s, 1H), 5.77 (brs, 2H), 3.87 (s, 3H).

Methyl 2-(4-tert-butylbenzamido)-4,5-dichlorobenzoate. To a microwave vial was added the methyl 2-amino-4,5-dichlorobenzoate (0.11 g, 0.48 mmol, 1 eq.), 4-tert-butylbenzoyl chloride (CAS# 1710-98-1) (0.096 mL, 0.527 mmol, 1.1 eq.) and MeCN (2.0 mL). The vial was sealed and the reaction stirred in a microwave at 150 °C for 30 minutes. The reaction was then cooled to rt and was diluted with EtOAc (10 mL) and washed with saturated NaHCO3 (10 mL). The crude material was then adsorbed onto silica. Purification by silica gel chromatography (0–15% EtOAc:Hex ramp over 10 min) afforded the desired product methyl 2-(4-tert-butylbenzamido)-4,5-dichlorobenzoate as a white solid (0.059 g, 0.16 mmol, 32% yield). 1H NMR (400 MHz, CDCl3) δ 11.94 (brs, 1H), 9.22 ( s, 1H), 8.15 (s, 1H), 7.96 (d, J = 8.6 Hz, 2H), 7.55 (d, J = 8.6 Hz, 2H), 3.99 (s, 3H), 1.36 (s, 9H).

ANALOGUE 3: 2-(4-tert-butylbenzamido)-4,5-dichlorobenzoic acid. (SID 99437306/CID 46947875) To a mixture of methyl 2-(4-tert-butylbenzamido)-4,5-dichlorobenzoate (0.019 g, 0.050 mmol, 1 eq.) and LiOH (0.010 g, 0.35 mmol, 7 eq.) in water (1 mL) was added THF (1 mL) to give a colorless solution. After stirring the solution for 3 h at rt, the reaction was acidified with 1.0 M aq. HCl to pH 2 – 3 and then extracted with CH2Cl2 (2 × 5 mL). The separated organic extracts were combined, dried (MgSO4), filtered and concentrated to produce the desired 2-(4-tert-butylbenzamido)-4,5-dichlorobenzoic acid as a white solid (0.017 g, 0.048 mmol, 96% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.17 (brs, 1H), 9.00 (s, 1H), 8.16 (s, 1H), 7.88 (d, J = 8.6 Hz, 2H), 7.63 (d, J = 8.6 Hz, 2H), 1.33 (s. 9H). 13C NMR (125 MHz, DMSO-d6) δ 168.2, 164.9, 155.7, 140.6, 136.4, 132.4, 131.0, 127.0, 126.0, 124.5, 121.1, 117.2, 34.8, 30.9. LCMS retention time: 2.514 min; Purity at 215 nm = 97.9%. HRMS m/z calculated for C18H17Cl2NO3 [M+ - 1] 365.06, found 364.05. White solid, mp 242 – 245 °C.
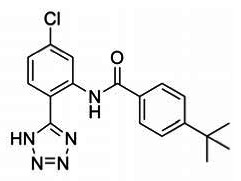
ANALOGUE 4: 4-(tert-butyl)-N-(5-chloro-2-(1H-tetrazol-5-yl)phenyl)benzamide (SID 99380773/CID 46916168): Isolated 67 mg, 61% yield of 4-(tert-butyl)-N-(5-chloro-2-(1H-tetrazol-5-yl)phenyl)benzamide as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.73 (s, 1H), 8.78 (d, J = 2.2 Hz, 1H), 8.04 (d, J = 8.5 Hz, 1H), 8.02 – 7.96 (m, 2H), 7.67 – 7.60 (m, 2H), 7.45 (dd, J = 8.5, 2.2 Hz, 1H), 1.34 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ 165.12, 155.47, 138.43, 136.25, 131.18, 129.98, 127.21, 125.77, 123.77, 120.63, 111.64, 97.71, 34.78, 30.83. LCMS retention time: 2.514 min; LCMS purity at 215 nm = 100%. HRMS m/z calculated for C18H18ClN5O [M++1]: 356.1273, found 356.1264. White solid, mp 231–234 °C.

Reagents: (a) NaN3, Et3N•HCl, PhMe, MWl, 110 °C
The requisite 5-chloro-2-(1H-tetrazol-5-yl)aniline precursor to analogue 4 (CID 46916168; SID 99380773) was prepared according to a previously reported procedure [15].

ANALOGUE 5: 2-(4-tert-butylbenzamido)-4-chloro-5-fluorobenzoic acid (SID 103023816/CID 49766619): This analogue was prepared according to the scheme shown below:

Reagents and conditions: a) acetic anhydride, acetic acid, reflux, 2 h; b) SOCl2, CH2Cl2, 45 °C for 2 h, 51% for two steps; c) 3-chloro-4-fluoroaniline, KHCO3, CH2Cl2, −10 °C - rt, 20 h; d) hydroxylamine-HCl, EtOH/H2O, reflux, 2 h, 45% for two steps; e) H2SO4, 80°C, 3 h; f) NaOH, H2O2, 80 °C, 3 h, 54% for two steps; g) TMS-diazomethane, CH2Cl2/MeOH, rt, 30 min, 82%; h) 4-tert-butylbenzoyl chloride, CH3CN, 150 °C, 50 min, MW, 56%; i) LiOH, THF/H2O, rt, 3 h, 94%.

2,2-diacetoxyacetyl chloride. The requisite starting materials glyoxylic acid monohydrate was purchased from Sigma-Aldrich (CAS # 563-96-2). To a vial was added glyoxylic acid monohydrate (2.05 g, 22.3 mmol, 1 eq.), acetic ahydride (21.0 mL, 223 mmol, 10 eq.) and acetic acid (5 mL). The colorless mixture was heated under reflux for 2 hr. The reaction was then cooled to rt, and the solvents were removed by rotary evaporation. The remaining volatiles were azeotroped with toluene and the crude material was solubilized in CH2Cl2. Thionyl chloride (5.68 mL, 78.1 mmol, 3.5 eq.) was added to the mixture and the reaction stirred at 45 °C for 2 hr. The reaction was cooled to rt, the volatiles were removed by rotary evaporation and the remaining material was isolated as the desired product, 2,2-diacetoxyacetyl chloride, as a brown oil (2.21 g, 11.4 mmol, 51% yield). 1H NMR (400 MHz, CDCl3) δ 6.89 (s, 1H), 2.19 (s, 6H).

N-(3-chloro-4-fluorophenyl)-2-(hydroxyimino)acetamide. The requisite starting material 3-chloro-4-fluoroaniline was purchased from Sigma-Aldrich (CAS # 367-21-5). To a vial was added the 3-chloro-4-fluoroaniline (2.71 g, 18.6 mmol, 1 eq.), potassium hydrogen-carbonate (9.31 g, 93.0 mmol, 5 eq.) in CH2Cl2 (35 mL), and the reaction was cooled to −10 °C. The 2,2-diacetoxyacetyl chloride (4.69 g, 24.2 mmol, 1.3 eq.) in CH2Cl2 (15 mL) was added dropwise and the reaction was allowed to warm to rt and stirred for 20 h. The reaction was filtered and rinsed with CH2Cl2 and the filtrate was concentrated. The crude material was then dissolved in EtOH:H2O (50 mL:20 mL) and hydroxylamine-HCl (6.46 g, 93.0 mmol, 5 eq.) was added. The reaction stirred at reflux for 2 hr, cooled to rt, and the EtOH was removed by rotary evaporation to form a precipitate. The solid was filtered and rinsed with water (50 mL) and dried on the filter to produce N-(3-chloro-4-fluorophenyl)-2-(hydroxyimino)acetamide as an off-white solid (1.83 g, 8.43 mmol, 45% yield). 1H NMR (400 MHz, CDCl3) δ12.25 (s, 1H), 10.38 (s, 1H), 7.99 (dd, J1 = 6.8 Hz, J2 = 2.6 Hz, 1H), 7.65 – 7.60 (m, 2H), 7.40 (t, J = 9.1 Hz, 1H).

2-Amino-4-chloro-5-fluorobenzoic acid. To a vial was added the N-(3-chloro-4-fluorophenyl)-2-(hydroxyimino)acetamide and concentrated sulfuric acid (30 mL). The reaction then stirred for 3 hr at 80°C, cooled to rt and poured onto ice. The product was extracted with CHCl3 (3 × 200 mL) and the organic layers were combined and concentrated by rotary evaporation. The crude material was solubilized in aqueous 2.5 M NaOH (20 mL), 30 % w/w hydrogen peroxide (2.50 mL, 24.3 mmol, 3 eq.) was added and the reaction stirred at 80°C for 3 h, cooled to rt and was acidified carefully with concentrated HCl to pH 3. The resulting precipitate was filtered and rinsed with water to produce 2-amino-4-chloro-5-fluorobenzoic acid as a reddish-brown solid (0.86 g, 4.60 mmol, 54% yield). 1H NMR (400 MHz, DMSO-d6) δ7.55 (d, J = 10.3 Hz, 1H), 6.93 (d, J = 6.5 Hz, 2H).
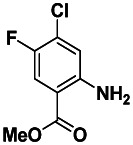
Methyl 2-amino-4-chloro-5-fluorobenzoate. To a vial was added 2-amino-4-chloro-5-fluorobenzoic acid (0.046 g, 0.24 mmol, 1 eq.) and CH2Cl2/MeOH (3 mL/2 mL). A solution of 2.0 M TMS-diazomethane in hexanes (0.22 mL, 0.44 mmol, 1.8 eq.) was added dropwise, and the reaction stirred for 30 minutes at rt. The crude material was then adsorbed onto silica. Purification by silica gel chromatography (0–20% EtOAc:Hex ramp over 20 min) afforded the desired product methyl 2-amino-4-chloro-5-fluorobenzoate as a white solid (0.041 g, 0.20 mmol, 82 % yield). 1H NMR (400 MHz, CDCl3) δ7.62 (d, J = 9.8 Hz, 1H), 6.71 (d, J = 6.1 Hz, 1H), 3.87 (s, 3H).

Methyl 2-(4-tertbutylbenzamido)-4-chloro-5-fluorobenzoate. To a microwave vial was added the methyl 2-amino-4-chloro-5-fluorobenzoate (0.076 g, 0.37 mmol, 1 eq.), 4-tert-butylbenzoyl chloride (CAS# 1710-98-1) (0.082 mL, 0.45 mmol, 1.2 eq.) and MeCN (2.0 mL). The vial was sealed and the reaction stirred in a microwave at 150 °C for 50 minutes. The reaction was then cooled to rt and was diluted with EtOAc (10 mL) and washed with saturated aqueous NaHCO3 (10 mL). The crude material was then adsorbed onto silica. Purification by silica gel chromatography (0–15% EtOAc:Hex ramp over 10 min) afforded the desired product methyl 2-(4-tertbutylbenzamido)-4-chloro-5-fluorobenzoate as a white solid (0.076 g, 0.21 mmol, 56% yield). 1H NMR (400 MHz, CDCl3) δ 11.89 (s, 1H), 9.17 (d, J = 7.1 Hz, 1H), 7.96 (d, J = 8.7 Hz, 2H), 7.84 (d, J = 9.5 Hz, 1H), 7.55 (d, J = 8.6 Hz, 2H), 3.98 (s, 3H), 1.36 (s, 9H).
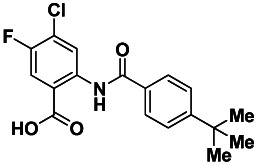
ANALOGUE 5: 2-(4-tert-butylbenzamido)-4-chloro-5-fluorobenzoic acid (SID 103023816/CID 49766619): To a mixture of methyl 2-(4-tertbutylbenzamido)-4-chloro-5-fluorobenzoate (0.076 g, 0.21 mmol, 1 eq.), and LiOH (0.035 g, 1.5 mmol, 7 eq.) in water (2 mL) was added THF (2 mL) to give a colorless solution. After stirring the solution for 3 h at rt, the reaction was acidified with 1.0 M aq. HCl to pH 2 – 3 and then extracted with CH2Cl2 (2 × 5 mL). The separated organic extracts were combined, dried (MgSO4), filtered and concentrated to produce the desired 2-(4-tertbutylbenzamido)-4-chloro-5-fluorobenzoic acid as a white solid (0.069 g, 0.196 mmol, 94% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 8.92 (d, J = 7.08 Hz, 1H), 7.96 (d, J = 9.8 Hz, 1H), 7.88 (d, J = 8.6 Hz, 2H), 7.62 (d, J = 8.6 Hz, 2H), 1.33 (s, 9H). 13C NMR (125 MHz, DMSO-d6) δ 168.3, 164.7, 155.5, 152.0 (d, J = 244.1 Hz), 138.1 (d, J = 2.8 Hz), 131.1, 127.0, 125.9, 125.0 (d, J = 18.1 Hz), 121.4, 118.5 (d, J = 23.0 Hz), 117.3 (d, J = 5.6 Hz), 34.8, 30.9. LCMS retention time = 3.641 min; purity at 215 nm = 98.3%. HRMS m/z calculated for C18H17ClFNO3 [M+ - 1] 349.09, found 350.09. White solid, mp 250 – 252 °C.
3. Results
The HTS effort produced three distinct scaffolds for possible development. Each of these was pursued with varying results, described in more detail below. However, one scaffold was amenable to optimization and was evaluated according to the flow chart in Figure 11. Based on knowledge of prior art, probe criteria was established as a compound showing rTbHK1 inhibition of ≤ 1 μM and not interfering with the reporter enzyme G6PDH (IC50 > 10x TbHK1 IC50). During the course of the project, the team agreed that assessment of the compounds in an IMR90 growth inhibition assay would be valuable, and this criterion was added to those desirable probe characteristics. Additionally, the team also decided to test compounds for potential undesirable cross reactivity, due to inhibition of the related human enzyme, hGlk. While not critical to the probe criteria, the assay provider also agreed to assess compounds in an additional assay for informative purposes to enhance the impact of the probe. Compounds were for evaluated for their ability to inhibit cell growth of T. brucei parasites in the bloodstream form cytotoxicity assay.
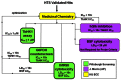
Figure 11
Triage of HTS hits and progression to meet probe criteria.
3.1. Summary of Screening Results
The Pittsburgh Molecular Libraries Screening Center (PMLSC, MLSCN phase) screened 220,223 compounds, available as part of the NIH Molecular Libraries Roadmap Initiative, at a single concentration (10 μM) for small molecule inhibitors of TbHK1 (AID 1430). The TbHK1 coupled assay was optimized and validated for HTS by screening the LOPAC set. Compounds were assayed in duplicate at a single concentration (10 μM) and reproducibility between the duplicate screens is represented in Figure 12A (R2 = 0.96). The HTS assay performed robustly (average Z-factors of 0.80 ± 0.1) and identified 239 compounds as primary actives (> 50% inhibition at 10 μM), for an overall hit rate of 0.1% (Fig 12B). The 239 active compounds were cherry-picked from DPI (of which 212 were available), and the initial inhibitory activity was confirmed in the primary TbHK1 assay (AID 2560) to afford 29 confirmed compounds. The compounds were then tested against the reporter enzyme, G6PDH, to verify that they did not interfere with the assay format (AID 2516). This effort afforded 16 hit compounds that were then assessed via a 20 point IC50 value determination (AID 1632) [7]. These assays are bundled in PubChem under summary AID 2600.

Of the 16 hits obtained from the HTS effort, three scaffolds were suitable from the perspective of lacking reactive functionality, being synthetically accessible, and demonstrating a reasonable activity profile (Figure 13).

Figure 13
HTS scaffolds and associated data prior to validation from resynthesized solid materials.
The two sulfonylbenzoates were resynthesized and upon retest, both compounds failed to confirm. Representatives from the isobenzothiazolinones and the one benzamidobenzoic acid were validated from resynthesized powder material.
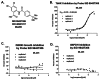
3.2. Dose Response Curves for Probe ML205, SID 99437306, CID 46931017
The dose response curves for probe ML205, SID 99437306, CID 46931017 are depicted in Figure 14. The probe demonstrated submicromolar activity versus rTbHK1 (IC50 = 0.98 μM, Fig. 14B). Probe ML205 (SID 99437306) did not show activity in the IMR90 growth inhibition assay or the G6PDH counterscreen (> 25 μM for both, Figure 14C and 14D, respectively).
3.3. Scaffold/Moiety Chemical Liabilities
The benzamidobenzoic acid scaffold and its derivatives have been easily handled in terms of stability to reaction conditions, exposure to acid or base, heating, and general manipulation. Most are isolated as stable solid materials. We have not observed decomposition nor have we experienced any chemical liability with these compounds. The structure does not contain moieties that are known generally to be reactive. The stability data that was collected has shown that after 48 hrs, 100% of the starting material remains (see section 2.2D, Figure 7).

Figure 7
Stability data depicted as a graph showing the loss of ML205 with time over a 48 hr period. The probe appears to be stable under the experimental conditions, as the percent of compound remaining was 100%.
3.4. SAR Tables and Discussion
Incipient SAR was observed from the initial HTS set of isobenzothiazolinones (see Figure 13), so a chemistry effort was launched around this primary scaffold (SID 17387000/CID 164981). Chemistry was developed to support the synthesis of compounds, and a total of 86 compounds were submitted for testing related to SID 17387000. The only clear trend observed with this chemotype was that the presence of the sulfur was necessary for any TbHK1 in vitro activity, thus implicating its participation in possible covalent interactions with nucleophilic residues, as we have had experience with this particular chemotype in the context of other projects and shown this to be true. While compounds were identified in the series with improved in vitro TbHK1 activity (SID 85285422, TbHK1 IC50 = 1.03 ± 0.17 μM) and lacking G6PDH activity (IC50 > 25 μM), clear SAR trends were difficult to draw out after a rigorous structural modification effort.
These results prompted the team to pursue the singleton benzamidobenzoic acid as a secondary chemotype. Over the course of the pursuing analogs of SID 24798131/CID 1254149, the parent hit has been tested 9 times in triplicate, demonstrating consistent in vitro TbHK1 activity on average of IC50 = 9.12 μM (n = 27) and a lack of activity in the G6PDH counterscreen, IC50 > 25 μM (n = 27). The chemotype was aggressively modified in the shaded regions of the diagram as part of our SAR strategy. A total of 102 compounds have been tested to date that relate to this scaffold. Of these, 83 were synthesized and 19 were purchased. All compounds, regardless of source, were purified and analyzed by LCMS and NMR for structural integrity and purity prior to assessment in assays.

A. Carboxylic acid isosteres
In most cases, replacement of the carboxylic acid with traditional isosteres resulted in loss of TbHK1 potency. Primary amides, sulfonamides, esters, or the use of an oxadiazolone were incorporated to mimic the hydrogen bonding and acidic nature of the carboxylic acid; however, all of these were found to be inferior to the parent. Installation of a nitro group, nitrile, a hydroxyl group or hydrogen at that position also resulted in no observable potency on TbHK1. Comparable activity to the carboxylic acid was achieved on substitution with a tetrazole, resulting in a TbHK1 IC50 = 10.4 μM (SID 99344412/CID 16124577). All compounds in this set did not interfere in the G6PDH counterscreen and possessed EC50s in the IMR90 cytotoxicity assay of > 25 μM (Table 2).
Table 2
SAR summary for compounds with carboxylic acid isosteric replacements.
B. Benzoic Acid Substitution
The 4-chlorophenyl substituent was targeted as a means of modulating the pKa of the benzoic acid and assessing the effect of different groups on the ring. Exchange of the 4-chloro group with 4-bromine was beneficial, resulting in a slight improvement in TbHK1 potency as compared to the parent (entry 2, SID 97302149/CID 46245547, IC50 = 6.9 μM); however other halides (entries 3–4), electron withdrawing or donating substituents (entries 5–6), larger substituents such as phenyl (entry 8), simple methyl substitution at the 4-position (entry 9), including removal of the 4-chloro group altogether (entry 10), resulted in erosion or complete loss of TbHK1 potency (Table 3). Moving the 4-chloro group to the 5-position was also not tolerated (entry 7). The effect of introducing other substituents on this ring while retaining the 4-chloro group was also surveyed. The introduction of a bromine at the 5-position notably improved potency (entry 12, IC50 = 4.6 μM) as compared to the parent (entry 1) while the installation of a 5-methyl group (entry 13) resulted in a marginal loss in potency. Interestingly, the 5 position appears to tolerate some substitution in concert with the 4-chloro group and may represent an opportunity to further refine the potency profile of this scaffold. Compounds with substitutions at either the 3- and 6- positions with the 4-chloro group retained are being prepared, as the 4-substituent has been determined to be critical to maintaining reasonable TbHK1 potency, possibly as a function of both size and electronics, and at present, some advantage has been made by functionalizing alternative positions (C3, C5 or C6). All completed compounds in this set did not interfere in the G6PDH counterscreen and possessed EC50s in the IMR90 cytotoxicity assay of > 25 μM.
Table 3
SAR summary for substitution on the benzoic acid moiety.
C. Amide Linker Modifications
The amide linker portion of the scaffold was particularly sensitive to modification. Any attempt to methylate the nitrogen (entry 2, Table 4), migrate the amide and associated appendage to the 3-position of the benzoic acid moiety (entry 5), truncate or elongate the linker region (entries 3, 6–7), or replace the amide with a sulfonamide resulted in complete loss of TbHK1 potency.
Table 4
SAR summary for substitution of the amide linker region.
Other analogs in this set that were pursued included compounds that featured a fused amide moiety (Figure 15). Fusion of the amide either to the methoxyphenyl appendage or the benzoic acid region was not tolerated.
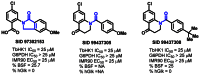
Figure 15
Fused amide analogs and associated data.
This data suggests, in concert with the data collected for carboxylic acid isosteric replacements in Table 2 and 3-substitution on the benzoic acid moiety (see Table 3, entry 11), that interference with hydrogen bonding (possibly between the acid functionality and the amide N-H) is detrimental.
D. Amidoaryl Modifications
The parental 4-methoxyphenyl moiety (parent: SID 24798131/CID 1254149) was probed extensively through structural variation to determine if any potency could be gained on TbHK1 as compared to the hit compound (Table 5). Migration of the 4-methoxy group to either the 3- or 2-positions on the ring resulted in progressively worse potencies, respectively. Comparable potency to that of the parent was obtained with a benzodioxole group (entry 7, TbHK1 IC50 = 9.4 μM); however, the similarly substituted 3,4-dimethoxyphenyl derivative (entry 6) demonstrated nearly a 2-fold loss in potency.
Table 5
SAR summary for modification of the amidoaryl component.
A series of analogues were also prepared that replaced the 4-methoxyphenyl group with various heterocycles or non-aromatic, cyclic entities that positioned polar functionality in the region where the 4-methoxy substituent of the parent resides (entries 9–17). In all cases, complete loss of TbHK1 activity was observed. Analogously, replacement of the complete methoxyphenyl moiety with linear aliphatic units was not tolerated (entries 19–23). Simple alterations (4-methoxy group → 4-Cl, 4-CF3, or 4-H) were also inferior.
Guided by the fact that polar functionality occupying similar space to the 4-methoxy substituent was undesirable, the team pursued analogues bearing lipophilic groups in that region of space. The 4-ethylphenyl derivative was the first of these that produced a 3-fold boost in potency (SID 25734182/CID 97302135, entry 34, TbHK1 IC50 = 3.0 μM). This region of space was further characterized with several analogues bearing branched or cyclic alkyl groups at the 3- or 4-positions (entries 32–42).
Migration of branched groups to the 3-position of the ring, again, resulted in loss of observable TbHK1 activity (> 25 μM); however, a few analogues were identified with improved TbHK1 activity when the 4-substituent was modified, specifically SID 99222689/CID 883362 (R = 4-t-butylphenyl, IC50 = 2.1 μM) and SID 99344415/CID 46891921 (R = 4-t-pentylphenyl, IC50 = 2.4 μM). Clearly, the data suggests that small, branched, lipophilic groups off of C4 are advantageous. That binding region also appears to concurrently accommodate some small lipophilic group off of C3, as evidenced by a few ring-fused analogs that occupy C3 and C4 simultaneously (entries 43–45). For these compounds, there was some modest compromise in TbHK1 activity, but these changes were still well-tolerated (IC50 range = 4.8 – 5.4 μM). A handful of analogs further exploring this region of space are currently in development to see if any enhancement of potency can be achieved. All compounds surveyed to date did not interfere in the G6PDH counterscreen and possessed EC50s in the IMR90 cytotoxicity assay of > 25 μM.
E. Tandem Modifications
Those structural features found to improve TbHk1 activity were combined in anticipation of a synergistic effect (Table 6). For instance, exchange of the chlorine of the parent (entry 1) for a bromine was determined to be acceptable, as was the optimization of the amidoaryl moiety with branched alkyl groups off of the 4-position. In the course of our investigation, the bromine and 4-ethylphenyl or 4-tert-butylphenyl combination were studied (entries 2 and 3), affording an improved TbHK1 activity of IC50 = 5.5 μM and 2.4 μM, respectively. As it has been shown, the best amidoaryl 4-substituent is a t-butyl group. This alteration was coupled with other features such as the tetrazole (entry 4), which was tolerated in place of the carboxylic acid. Additionally, the substitution pattern of the benzoic acid moiety of R1 = Cl, R2 = Br (entry 7) was discovered to be the most beneficial optimization, resulting in a submicromolar inhibition of TbHK1 and culminating in the declaration of a probe compound for this project.
Table 6
Survey of combined effects of optimized functionality.
3.5. Cellular Activity
Compounds were evaluated in a human IMR90 growth inhibition assay (AID 449725) to assess mammalian cytotoxicity. All compounds in the series possessed EC50s in the IMR90 cytotoxicity assay of > 25 μM. Data from the whole parasite BSF assay, while not required for probe criteria, was collected for many of the compounds in this series. Most compounds were assessed at a single concentration of 10 μM for percent growth inhibition of parasites and all registered in the range of 0–55%. The SAR profile relating to the percent BSF inhibition is not quite clear at this point in time; however, the compounds do show toxicity towards parasites to varying degrees. Select examples are shown in Figure 16 and in the preceding SAR tables.
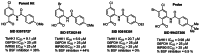
Figure 16
Select examples of cellular IMR90 and percent BSF inhibition data.
3.6. Profiling Assays
The probe compound ML205, SID 99437306, CID 46931017 and the purified prior art compound quercetin (SID 99460891/CID 5280343) were profiled at 5 μM in a 50-member kinase panel to assess differences in selectivity between the two compounds [16].
| Kinase | Family | PROBE | QUERCETIN |
|---|---|---|---|
| % Activity Remaining SID 99437306 | % Activity Remaining SID 99460891 | ||
| AKT1(FL) | AGC | 100 | 100 |
| AKT2 | AGC | 99.4 | 100 |
| AMPK-Ą1 | CAMK | 82.2 | 100 |
| AURKA | Other | 98.8 | 97.9 |
| AURKB | Other | 100 | 89.4 |
| AURKC | Other | 100 | 100 |
| BLK | TK | 96.5 | 95.7 |
| CAMK1 | CAMK | 100 | 100 |
| CAMK1D | CAMK | 91.3 | 100 |
| CAMKK1 | Other | 98.4 | 100 |
| CAMKK2 | Other | 84.8 | 100 |
| CHEK1 | CAMK | 100 | 100 |
| CLK1 | CMGC | 92.8 | 70.3 |
| CLK2 | CMGC | 96.8 | 100 |
| DDR2 | TK | 73.1 | 100 |
| FGFR2 | TK | 89.4 | 100 |
| FLT1 | TK | 97.4 | 100 |
| FYN | TK | 100 | 100 |
| GSK3Ą | CMGC | 100 | 92.1 |
| IGF1R | TK | 100 | 100 |
| ITK | TK | 100 | 100 |
| LYN | TK | 100 | 100 |
| MARK1 | CAMK | 66.9 | 100 |
| Met | TK | 100 | 100 |
| MLK1 | TKL | 100 | 98.6 |
| MLK3 | TKL | 87.4 | 100 |
| MST2 | STE | 100 | 100 |
| p38-ƒ× | CMGC | 100 | 100 |
| PAK1 | STE | 100 | 100 |
| PDGFRB | TK | 100 | 80 |
| PDK1 | AGC | 100 | 100 |
| PIM1 | CAMK | 80.5 | 34.3 |
| PIM2 | CAMK | 100 | 100 |
| PKAC-Ą | AGC | 88.8 | 100 |
| PKC-ƒ× | AGC | 100 | 100 |
| PRKD2 | CAMK | 100 | 100 |
| PKG1 | AGC | 98 | 100 |
| PLK4 | Other | 100 | 100 |
| PTK2 | TK | 100 | 100 |
| RPS6KA1 | AGC | 100 | 100 |
| SNF1LK | CAMK | 100 | 100 |
| SLK | STE | 100 | 100 |
| SNARK | CAMK | 100 | 100 |
| SRC | TK | 100 | 100 |
| SRPK2 | CMGC | 100 | 100 |
| SYK | TK | 100 | 100 |
| TNK2 | TK | 100 | 100 |
| VEGFR2 | TK | 100 | 96.8 |
| YES1 | TK | 100 | 100 |
| YSK1 | STE | 100 | 100 |
For most of the kinases assayed, both the probe and quercetin showed relatively low (0–10%) inhibition. In the case of PIM1, quercetin showed significant inhibition (~66%) while the probe compound demonstrated inhibition at ~ 20%. Profiling data for all kinases was plotted as percent activity remaining vs. kinases profiled, represented below:

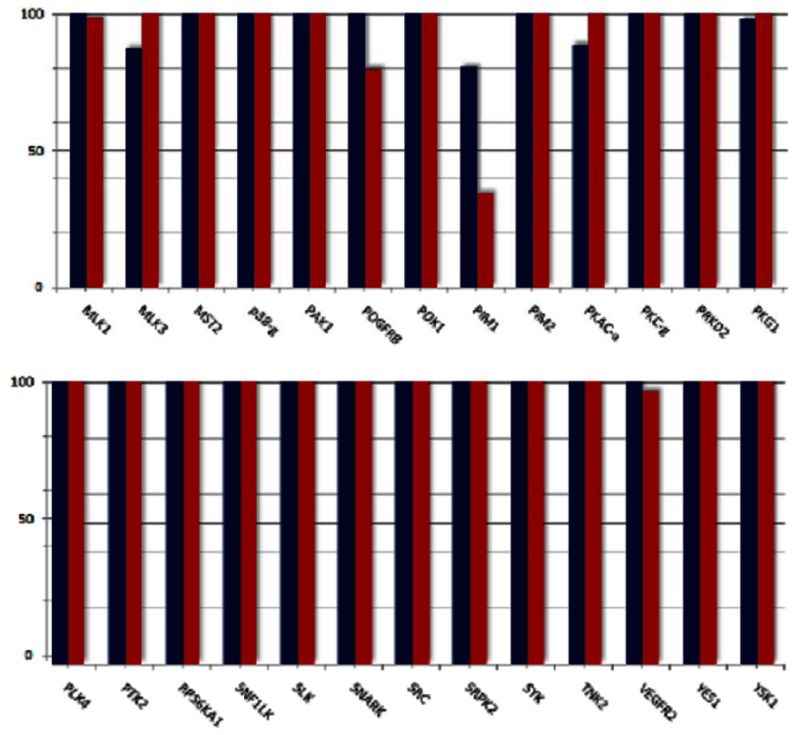
Human glucokinase was not available as part of the panel, but the Assay Provider assessed compounds for this target at 10 μM (AID 492951). The probe compound did not significantly inhibit hGlk (IC50 = 48.3 μM) while quercetin inhibited the same target with an IC50 of 2.2 μM.


4. Discussion
4.1. Comparison to existing art and how the new probe is an improvement
The HTS effort that identified a viable hit scaffold and the optimization of the parent hit SID 24798131/CID 1254149 with rTbHK1 IC50 = 9.1 μM to a probe that inhibits rTbHK1 in the submicromolar range is a significant achievement given the limited leads and therapeutics available. This project has delivered a probe that meets all of the criteria set forth in the CPDP and significantly improves upon prior art in the (1) potency on the in vitro rTbHK1 target, (2) structural improvements to avoid reactive functionality, and (3) better selectivity against hGlk (a related human kinase) and PIM1 (important in cell cycle regulation and apoposis). Given that quercetin has been shown to inhibit a variety of targets, thus limiting its use with respect to definitive hexokinase pathway investigation, the increased value of the probe defined here is in its ability to avoid these off-target liabilities [8].
Table 7Comparison of quercetin and probe
| Parameter or Biological Characteristics | Desired Probe Characteristic | Quercetin | Probe ML205 | Probe meets criteria? |
|---|---|---|---|---|
| SID | NA | 99460891 | 99437306 | NA |
| CID | NA | 5280343 | 46931017 | NA |
| rTbHK1 potency (IC50, nM) | IC50 ≤ 1000 nM | IC50 = 22400 nM | IC50 = 976 nM | Yes ✓ |
| selectivity vs G6PDH reporter enzyme (nM) | IC50 ≥ 25000 nM | IC50 > 25000 nM | IC50 > 25000 nM | Yes ✓ |
| Biological Mode of Action/Assessment | in vitro | in vitro | in vitro | Yes ✓ |
| Pharmacology/Mode of Action (allosteric/covalent, etc) | mixed inhibition with respect to ATP | mixed inhibition with respect to ATP | mixed inhibition with respect to ATP | Yes ✓ |
| Cellular Toxicity in IMR90 cells | EC50 > 25000 nM (or 10x BSF LD50) | EC50 > 25000 nM | EC50 > 25000 nM | Yes ✓ |
| Structural Features to Avoid | Non-flavinol | Flavinol | Non-flavinol | Yes ✓ |
| selectivity vs human glucokinase (hGlk) % inhibition at 10 μM or IC50 | IC50 > 10x TbHK1 IC50 | IC50 = 2200 nM (nonselective) | IC50 = 48300 nM | Yes ✓ |
| Cytotoxicity towards whole BSF parasites (% inhibition at 10 μM or LD50 ) - not required | LD50 < 25000 nM | LD50 = 2950 nM | % inhibition at 10 μM = 6.9 | Not required |
| off target liability - not required | selective against a panel of kinases | promiscuous against various kinases, specifically hGlk and PIM1 | selective against a panel of kinases | Not required |
4.2. Mechanism of Action Studies
In an attempt to better understand the nature of the inhibition associated with these compounds on TbHK1, an additional study was completed by the assay provider, Dr. James Morris (Fig. 17). Preliminary studies suggest that SID 99437306/CID 46931017 is a mixed inhibitor of TbHK1 with respect to ATP with a Ki of 0.695 μM. This is notable because screens for inhibitors of kinases have tended to yield competitive inhibitors that are frequently ATP analogues – SID 99437306/CID 46931017 may represent a novel, non-ATP binding site scaffold for probe improvement against TbHK1.

Figure 17
Michaelis-Menten and Lineweaver-Burk plots showing mixed inhibition of TbHK1 with respect to ATP using ML205, SID 99437306, CID 46931017.
4.3. Planned Future Studies
A probe advancement proposal was submitted to the NIH as of October 13, 2010, and has been subsequently approved. While some regions of the scaffold represented by probe ML205 were not amenable to SAR modification without adversely affecting potency, optimization was possible in four sectors that were investigated. There remain a few diversification points which were not included in this effort which may lead to improved profile and opportunity to refine physiochemical parameters. The following specific SAR plans are being implemented to address potency and cellular permeability.

Figure 18Planned SAR optimization for extended probe characterization
The areas highlighted above will be investigated to meet the goals of the project. Unaddressed areas of the structure are not being modified as these were extensively studied in the probe project leading to the development of ML205; however, these regions may be altered in the event that structural variation elsewhere gains sufficient activity. In the probe enhancement phase, the probe will be compared to quercetin and assessed in the following ways:
| Parameter or Biological Characteristics | Quercetin | Probe ML205 | Desired Optimized Probe |
|---|---|---|---|
| SID | 99460891 | 99437306 | NA |
| CID | 5280343 | 46931017 | NA |
| rTbHK1 potency (IC50, nM) | IC50 = 22400 nM | IC50 = 976 nM | IC50 ≤ 500 nM |
| rTbHK1 potency (IC50, nM) in ATP- regeneration coupled assay | unknown | unknown | IC50 ≤ 500 nM |
| Inhibit TbHK1 activity from T. brucei parasite lysate | IC50 = 24000 nM** | unknown | IC50 ≤ 500 nM |
| Cytotoxicity towards whole BSF parasites (% inhibition at 10 μM or LD50) | LD50 = 2950 nM | % inhibition at 10 μM = 6.9 | LD50 of ≤ 1000 nM |
| Confirmation of action on the in vivo target, as determined by measuring the impact of compound on parasitic cellular G6P levels | unknown | unknown | > 30% |
| Selectivity vs yeast hexokinase | unknown | unknown | ≤ 25% inhibition or IC50 > 10,000 nM |
| Selectivity vs human hexokinase 1 | unknown | unknown | < 25% inhibition or IC50 > 10,000 nM |
| selectivity vs human glucokinase (hGlk) % inhibition at 10 μM or IC50 | IC50 = 2200 nM (nonselective) | IC50 = 48300 nM | < 50% inhibition at 10 μM; IC50 > 10x TbHK1 IC50 |
| selectivity vs G6PDH reporter enzyme (nM) | IC50 > 25000 nM | IC50 > 25000 nM | IC50 > 25000 nM |
| limited other off target liability (Luceome profiling) | promiscuous against various kinases, specifically hGlk and PIM1 | selective against a panel of kinases | selective against a panel of kinases |
| Demonstrate efficacy in the Leishmania promastigote assay | unknown | unknown | informative, but not critical to probe status |
| Biological Mode of Action/Assessment | in vitro | in vitro | in vivo |
| Pharmacology/Mode of Action (allosteric/covalent, etc) | mixed inhibition with respect to ATP | mixed inhibition with respect to ATP | mixed inhibition with respect to ATP |
| Cellular Toxicity in IMR90 cells | EC50 > 25000 nM | EC50 > 25000 nM | EC50 > 25000 nM (or 10x BSF LD50) |
| Aqueous solubility 1x PBS buffer (ug/mL) pH 7.4 | unknown | 25.6 ug/mL | ≥ 50 ug/mL |
| Aqueous stability (% remaining after 48 h) | unknown | 100% | ≥ 95% |
| cellular permeability (10−6 cm/s ) | unknown | unknown | medium-high |
| microsomal stability t1/2(mouse) [min] | unknown | unknown | 20–40 min* |
| plasma protein binding | unknown | unknown | < 97%* |
| plasma stability | unknown | unknown | > 95%* |
| efficacy in acute infection mouse model | unknown | unknown | TBD mg/kg |
- *
These values are provided as a guideline, but it should be noted that these can be highly variable and depend on pharmacokinetics as a whole. As a result, a compound may have acceptable PK due to the interplay of these characteristics while the individual characteristic may fall out of the desired guideline.
- **
TbHK activity (may be due to either TbHK1 or TbHK2)
Compounds will be provided by the KU SCC in sufficient quantities (500 mg – 1 gram) and purity for the proposed studies and will likely be further refined structurally to tune physiochemical and pharmacokinetic parameters necessary for adequate analysis in rodent models. Specifically, the probe will be assessed by the KU SCC and structurally optimized to:
- possess suitable aqueous solubility, stability, and cellular permeability
- possess reasonable microsomal stability, plasma protein binding and plasma stability
- be assessed for in vivo pharmacokinetics
5. References
- 1.
- WHO Media centre. Fact sheet N°259: African trypanosomiasis or sleeping sickness. 2010. http://www
.who.int/mediacentre /factsheets/fs259/en/ - 2.
- Remme JH, Blas E, Chitsulo L, Desjeux PM, Engers HD, et al. Strategic emphases for tropical diseases research: a TDR perspective. Trends Parasitol. 2002;18:421–426. [PubMed: 12377584]
- 3.
- Jacobs RT, Ding C. Recent advances in drug discovery for neglected tropical diseases casued by infective kinetoplastid parasites. Annual Reports in Medicinal Chemistry. 2010;45:277–294.
- 4.
- Trinquier M, Perie J, Callens M, Opperdoes F, Willson M. Specific inhibitors for the glycolytic enzymes of Trypanosoma brucei. Bioorg. Med. Chem. 1995;3:1423–1427. [PubMed: 8634823]
- 5.
- Willson M, Sanejouand YH, Perie J, Hannaert V, Opperdoes F. Sequencing, modeling, and selective inhibition of Trypanosoma brucei hexokinase. Chem Biol. 2002;9:839–847. [PubMed: 12144928]
- 6.
- Chambers JW, Fowler ML, Morris MT, Morris JC. The antitrypanosomal agent lonidamine inhibits Trypanosoma brucei hexokinase 1. Mol Biochem Parasitol. 2008;158:202–207. [PubMed: 18262292]
- 7.
- Sharlow ER, Lyda TA, Dodson HC, Mustata G, Morris MT, Leimgruber SS, Lee K-H, Kashiwada Y, Close D, Lazo JS, Morris JC. A target-based high throughput screen yields Trypanosoma brucei hexokinase small molecule inhibitors with antiparasitic activity. PLoS Negl Trop Dis. 2010;4:e659. (7 pages) [PMC free article: PMC2854128] [PubMed: 20405000]
- 8.
- Dodson HC, Lyda TL, Chambers JW, Morris MT, Christensen KA, Morris JC. Quercetin, a fluorescent bioflavanoid, inhibits Trypanosoma brucei hexokinase 1. Experimental Parasitology. 2010 In press. [PMC free article: PMC3025057] [PubMed: 20971104]
- 9.
- Srivastava AK. Inhibition of phosphorylase kinase, and tyrosine protein kinase activities by quercetin. Biochem Biophy Res Commun. 1985;131:1–5. [PubMed: 4041183]
- 10.
- Matter WF, Brown RF, Vlahos CJ. The inhibition of phosphatidylinositol 3-kinase by quercetin and analogs. Biochem Biophy Res Commun. 1992;186:624–631. [PubMed: 1323287]
- 11.
- Boege F, Straub T, Kehr A, Boesenberg C, Christiansen K, Andersen A, Jakob F, Kohrle J. Selected novel flavones inhibit the DNA binding or the DNA religation step of eukaryotic topoisomerase I. J Biol Chem. 1996;271:2262–2270. [PubMed: 8567688]
- 12.
- Solubility and stability data was outsourced to and collected by the Sanford-Burnham Center, Dr. Layton Smith
- 13.
- Dreher SD, Dormer PG, Sandrock DL, Molander GA. Efficient cross-coupling of secondary alkyltrifluoroborates with aryl chlorides - reaction discovery using parallel microscale experimentation. J Am Chem Soc. 2008;130:9257–9259. [PMC free article: PMC2593853] [PubMed: 18582050]
- 14.
- Molander GA, Gormisky PE. Cross-coupling of cyclopropyl- and cyclobutyltrifluoroborates with aryl and heteroaryl chlorides. J Org Chem. 2008;73:7481–7485. [PMC free article: PMC2635095] [PubMed: 18759480]
- 15.
- Koguro K, Oga T, Mitsui S, Orita R. Novel synthesis of 5-substituted tetrazoles from nitriles. Synthesis. 1998;6:910–914.
- 16.
- Data obtained from Luceome Biotechnologies using the KinaseSeeker™ assay. For information on the assay principle and method, please refer to: Jester, B. W.; Cox, K. J.; Gaj, A.; Shomin, C. D.; Porter, J. R.; Ghosh, I. "A Coiled Coil Enabled Split-Luciferase Three-Hybrid System: Applied Toward Profiling Inhibitors of Protein Kinases" J. Am. Chem. Soc. 2010, 132, 11727-11735. Assay design and method: Compounds were dissolved in DMSO and tested at a final concentration of 5 µM in all experiments. Prior to initiating a profiling campaign, the compound was evaluated for false positive against split-luciferase. After it was determined that it did not inhibit the luciferase control at 5 µM concentration, profiling was done in duplicate for against each kinase. The % Inhibition and % Activity Remaining was calculated using the following equation: % Inhibition = (ALUControl – ALUSample ) /ALUControl x 100; % Activity Remaining = 100 - % Inhibition.
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Review Identification of Selective Inhibitors of Phosphofructokinase as Lead Compounds Against Trypanosomiasis.[Probe Reports from the NIH Mol...]Review Identification of Selective Inhibitors of Phosphofructokinase as Lead Compounds Against Trypanosomiasis.Walsh MJ, Brimacombe KR, Vásquez-Valdivieso MG, Auld DS, Simeonov A, Morgan HP, Fothergill-Gilmore LA, Michels PAM, Walkinshaw MD, Shen M, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Development of drug resistance in Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense. Treatment of human African trypanosomiasis with natural products (Review).[Int J Mol Med. 2008]Review Development of drug resistance in Trypanosoma brucei rhodesiense and Trypanosoma brucei gambiense. Treatment of human African trypanosomiasis with natural products (Review).Gehrig S, Efferth T. Int J Mol Med. 2008 Oct; 22(4):411-9.
- Screening North American plant extracts in vitro against Trypanosoma brucei for discovery of new antitrypanosomal drug leads.[BMC Complement Altern Med. 2016]Screening North American plant extracts in vitro against Trypanosoma brucei for discovery of new antitrypanosomal drug leads.Jain S, Jacob M, Walker L, Tekwani B. BMC Complement Altern Med. 2016 May 18; 16:131. Epub 2016 May 18.
- 3-(Oxazolo[4,5-b]pyridin-2-yl)anilides as a novel class of potent inhibitors for the kinetoplastid Trypanosoma brucei, the causative agent for human African trypanosomiasis.[Eur J Med Chem. 2013]3-(Oxazolo[4,5-b]pyridin-2-yl)anilides as a novel class of potent inhibitors for the kinetoplastid Trypanosoma brucei, the causative agent for human African trypanosomiasis.Ferrins L, Rahmani R, Sykes ML, Jones AJ, Avery VM, Teston E, Almohaywi B, Yin J, Smith J, Hyland C, et al. Eur J Med Chem. 2013 Aug; 66:450-65. Epub 2013 May 16.
- Evaluating the impact of targeting livestock for the prevention of human and animal trypanosomiasis, at village level, in districts newly affected with T. b. rhodesiense in Uganda.[Infect Dis Poverty. 2017]Evaluating the impact of targeting livestock for the prevention of human and animal trypanosomiasis, at village level, in districts newly affected with T. b. rhodesiense in Uganda.Hamill L, Picozzi K, Fyfe J, von Wissmann B, Wastling S, Wardrop N, Selby R, Acup CA, Bardosh KL, Muhanguzi D, et al. Infect Dis Poverty. 2017 Feb 6; 6(1):16. Epub 2017 Feb 6.
- Identification of Inhibitors of Trypanosoma brucei Hexokinases - Probe Reports f...Identification of Inhibitors of Trypanosoma brucei Hexokinases - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...