

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

Optimization and characterization of a carbamate inhibitor for plasma platelet-activating factor acetylhydrolase (pPAFAH)
Joseph M G Nagano, Ku-Lung Hsu, Anna E Speers, Steven J Brown, Timothy Spicer, Virneliz Fernandez-Vega, Jill Ferguson, Brian J Bahnson, Benjamin F Cravatt, Peter Hodder, and Hugh Rosen.
Author Information and Affiliations
Received: November 30, 2011; Last Update: May 13, 2014.
Oxidative stress has been implicated as an underlying inflammatory factor in several disease pathologies, including cancer, atherosclerosis, aging, and various neurodegenerative disorders. Phospholipids in particular are susceptible to oxidative damage, and it is thought that the plasma platelet-activating factor acetylhydrolase (pPAFAH, a.k.a. PLA2G7) may facilitate turnover of oxidized phospholipids via hydrolysis of their oxidatively truncated acyl chains. However, there are no commercially available, selective, and in vivo-active inhibitors for investigation of pPAFAH biology. As such, we initiated a fluorescence polarization activity-based protein profiling (fluopol-ABPP) high throughput screening (HTS) campaign to identify potential inhibitors of pPAFAH and three PAFAH family members: PAFAH2, PAFAHb2, and PAFAH1b3.
The pPAFAH HTS campaign revealed a large number (~5000) of potential lead compounds, but secondary gel-based screening of ~150 cherry picked top inhibitors quickly revealed the carbamate as a promising scaffold for inhibitor design. Given that carbamate inhibitors for serine hydrolase enzymes have previously been found to have tunable potency and selectivity and good activity in vivo, we endeavored to derive a pPAFAH-selective probe from the carbamate scaffold. In this effort, we were aided by the recent identification of a lead pPAFAH carbamate inhibitor from a small in-house library. By combining elements of both the HTS carbamate hits and our in-house lead, the medchem optimized probe (ML256, SID 125269079) is highly potent against its target enzyme (IC50 = 31 nM mouse isoform; 6 nM human isoform), and is active in situ and in vivo, showing excellent oral bioavailability and blood-brain barrier penetration. ML256 is at least 322-fold selective for all other brain serine hydrolases (~20) assessed by gel-based competitive activity-based protein profiling (ABPP), and is selective for other PAFAH enzymes as determined by both gel-based screening and multidimensional LC-MS/MS analysis (ABPP-MudPIT). ML256 inhibits pPAFAH by irreversible carbamoylation of the active site serine. The complete properties, characterization, and synthesis of ML256 are detailed in this report.
Assigned Assay Grant #: 1 R01 HL084366
Screening Center Name & PI: The Scripps Research Institute Molecular Screening Center (SRIMSC), H Rosen
Chemistry Center Name & PI: SRIMSC, H Rosen
Assay Submitter & Institution: BJ Bahnson, Univ. of DE; BF Cravatt, TSRI, La Jolla
PubChem Summary Bioassay Identifier (AID): 463092
Probe Structure & Characteristics
| CID/ML# | Target Name | Target IC50 (nM) [SID, AID] | Anti-target Name(s) | Anti-target IC50 (nM) [SID, AID] | Fold Selective† | Secondary Assay(s) Name: IC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 53364507/ML256 | pPAFAH (Pla2g7) | Mouse: 31 [SID 125269079, AID 588770] Human: 6 [SID 125269079, AID 588766] | >20 brain SHs* | >10,000 [SID 125269079, AID 588774]** | >322 | Inhibition Assay: [SID 125269079, AID 588774] Selectivity Assay: [SID 125269079, AID 588774, AID 588767] MudPIT Selectivity Assay: [SID 125269079, AID 588785 and AID 588787] In Situ Assay: [SID 125269079, AID 588817] In Vivo Assay: [SID 125269079, AID 588823] IC50 Assay: mouse 31 nM [SID 125269079, AID 588770] ; human 6 nM [SID 125269079, AID 588766] Cytox assay: [SID 125269079, AID 588768] LC-MS/MS assay: [SID 125269079, AID 588788] |
| PAFAH2 (closest homolog) | >10,000 [SID 125269079, AID 588767]** | >322 |
- *
As assessed by gel-based competitive ABPP in the mouse brain membrane proteome with the serine hydrolase-specific activity-based probe FP-Rhodamine
- **
IC50 of the anti-target is defined as greater than the test compound concentration at which less than or equal to 50% inhibition of the anti-target is observed, which is reported in AID 588774. For SID 125269079, no anti-targets were observed for all serine hydrolases (SHs) assayed at 10,000 nM concentration, so the IC50 is reported as >10,000 nM.
- †
Fold-selectivity was calculated as: >IC50 for anti-target/IC50 for pPAFAH
Recommendations for scientific use of the probe
Oxidative stress has been implicated as an underlying inflammatory factor in numerous disease pathologies, including cancer, atherosclerosis, aging, and neurodegenerative disorders [1–5]. Phospholipids in particular are susceptible to oxidative damage, and it is thought that pPAFAH may facilitate turnover of oxidized phospholipids via hydrolysis of their oxidatively truncated acyl chains. ML256, described herein, is a potent and specific inhibitor of pPAFAH in vitro, in situ, and in vivo. As such, it is recommended for use in primary research studies aimed at elucidating the patho/physiological roles and substrate specificity of pPAFAH and its contribution to inflammatory disease processes.
1. Introduction
Oxidative stress has been implicated as an underlying inflammatory factor in several disease pathologies, including cancer, atherosclerosis, aging, and various neurodegenerative disorders [1–5]. Phospholipids in particular are susceptible to oxidative damage, and (per)oxidized phospholipids can have deleterious effects, including disruption of membrane bilayers and production of toxic byproducts [4, 6–8]. One hypothesized pathway for removal of oxidatively damaged lipids involves hydrolysis by phospholipase A2-type enzymes. Candidate hydrolytic enzymes include the platelet-activating factor acetylhydrolases (PAFAHs) [4, 9]. The initial role assigned to the PAFAHs was the hydrolysis of platelet activating factor (PAF) [10, 11], a potent pro-inflammatory phospholipid signaling molecule [12], which plays a role in myriad physiological processes including inflammation, anaphylaxis, fetal development, and reproduction [4, 13]. The PAFAHs are subdivided into three classes: plasma (p)PAFAH (a.k.a. PLA2G7), and intracellular types 1 and 2. In terms of sequence homology, pPAFAH and PAFAH2 are close homologs and show little similarity to type 1 enzymes.
A large number of studies have been published over the years since pPAFAH was first discovered linking an increase in pPAFAH concentration and/or activity to an increased risk of various cardiovascular diseases [14, 15]. The biological function of pPAFAH in the development of coronary heart diseases (CHD) is controversial, with both anti- and proinflammatory roles attributed to it [16, 17]. Chemical tools capable of interrogating enzyme architecture (co-crystallization studies) and providing precise temporal control over activity of pPAFAH and the other PAFAH-family enzymes are necessary for complete characterization of substrate specificity and patho/physiological roles of the PAFAHs in phospholipid metabolism and inflammatory disease processes. Towards that goal, we developed a HTS assay for inhibitor discovery for four PAFAH enzymes: pPAFAH, PAFAH2, PAFAH1b2, and PAFAH1b3, based on their reactivity with the serine-hydrolase-specific fluorophosphonate (FP) [18] activity-based protein profiling (ABPP) probe. This reactivity can be exploited for inhibitor discovery using a competitive-ABPP platform, whereby small molecule enzyme inhibition is assessed by the ability to out-compete ABPP probe labeling [19]. Competitive ABPP has also been configured to operate in a high-throughput manner via fluorescence polarization readout, fluopol-ABPP [20]. This HTS campaign followed by medchem optimization has already delivered an optimized triazole urea probe for PAFAH2, ML225. Following the fluopol-ABPP HTS campaign for pPAFAH and secondary gel-based competitive ABPP screening, we identified several carbamate hit compounds with IC50s of 200–400 μM. By combining structural elements of these compounds with an in-house carbamate lead, WWL153 (SID 99205832), we derived probe ML256 (SID 125269079), which is highly potent against pPAFAH (IC50 31 nM for the mouse isoform, 6 nM for the human isoform), and is active in situ and in vivo, inhibiting its target enzyme in the mouse brain following oral administration. ML256 is at least 322-fold selective for all other brain serine hydrolases (SHs) (~20) assessed by gel-based competitive ABPP, and is selective for the counter-screen enzymes pPAFAH, PAFAH1b2, and PAFAH1b3. ML256 inhibits its target by carbamoylating the active site serine. While reversible inhibitors (darapladib) of pPAFAH have been reported [21], these compounds are not commercially available and have not been extensively characterized in vivo due to the lack of known biomarkers to assess pPAFAH inhibition in living systems. ML256 is the first selective, covalent inhibitor of pPAFAH, and we show that its activity against pPAFAH can be evaluated in living systems, including mice, using competitive ABPP methods.
2. Materials and Methods
All reagents for chemical synthesis were obtained from ThermoFisher or SigmaAldrich. All other protocols are summarized in Section 2.1.
2.1. Assays
Probe Characterization Assays
Solubility
The solubility of compounds was tested in phosphate buffered saline, pH 7.4. Compounds were inverted for 24 hours in test tubes containing 1–2 mg of compound with 1 mL of PBS. The samples were centrifuged and analyzed by HPLC (Agilent 1100 with diode-array detector). Peak area was compared to a standard of known concentration.
Stability
Demonstration of stability in PBS was conducted under conditions likely to be experienced in a laboratory setting. The compound was dissolved in 1 mL of PBS at a concentration of 10 μM, unless its maximum solubility was insufficient to achieve this concentration. Low solubility compounds were tested between ten and fifty percent of their solubility limit. The solution was immediately aliquoted into seven standard polypropylene microcentrifuge tubes which were stored at ambient temperature in a block microcentrifuge tube holder. Individual tubes were frozen at −80°C at 0, 1, 2, 4, 8, 24, and 48 hours. The frozen samples were thawed in a room temperature and an equal volume of acetonitrile was added prior to determination of concentration by LC-MS/MS.
LC-MS/MS for stability assay
All analytical methods are in MRM mode where the parent ion is selected in Q1 of the mass spectrometer. The parent ion is fragmented and a characteristic fragment ion is monitored in Q3. MRM mass spectroscopy methods are particularly sensitive because additional time is spent monitoring the desired ions and not sweeping a large mass range. Methods will be rapidly set up using Automaton® (Applied Biosystems), where the compounds are listed with their name and mass in an Excel datasheet. Compounds are submitted in a 96-well plate to the HPLC autosampler and are slowly injected without a column present. A narrow range centered on the indicated mass is scanned to detect the parent ion. The software then evaluates a few pre-selected parameters to determine conditions that maximize the signal for the parent ion. The molecule is then fragmented in the collision cell of the mass spectrometer and fragments with m/z larger than 70 but smaller than the parent mass are determined. Three separate collision energies are evaluated to fragment the parent ion and the largest three ions are selected. Each of these three fragment ions is further optimized and the best fragment is chosen. The software then inserts the optimized masses and parameters into a template method and saves it with a unique name that indicates the individual compound being optimized. Spectra for the parent ion and the fragmentation pattern are saved and can be reviewed later.
Determination of glutathione reactivity
One μL of a 10 mM compound stock solution was added to 1 mL of a freshly prepared solution of 100 μM reduced glutathione. Final compound concentration is 10 μM unless limited by solubility. The solution was allowed to incubate at 37°C for two hours prior to being directly analyzed for glutathione adduct formation. LC-MS/MS analysis of GSH adducts was performed on an API 4000 Q-TrapTM mass spectrometer equipped with a Turboionspray source (Applied Biosystems, Foster City, CA). Two methodologies were utilized: a negative precursor ion (PI) scan of m/z 272, corresponding to GSH fragmenting at the thioether bond, and a neutral loss scan of −129 AMU to detect GSH adducts. This triggered positive ion enhanced resolution and enhanced product ion scans [22, 23].
Primary Assays
Primary uHTS assay to identify pPAFAH inhibitors (AID 463082)
Assay Overview: The purpose of this assay is to identify compounds that act as inhibitors of the plasma platelet activating factor acetylhydrolase (pPAFAH). In this biochemical assay, recombinant human pPAFAH protein (purified GST-fusion construct expressed in E. coli BL21 cells, contains residues 42–441 plus a non-native N-terminal extension [GSPNSRVD] and three mutations in the hydrophobic patch region: I120A, L123A, L124A) is incubated with test compounds for a defined period, followed by addition of a rhodamine-conjugated fluorophosphonate (FP-Rh) serine hydrolase-specific activity-based probe. The reaction is excited with linear polarized light and the intensity of the emitted light is measured as the polarization value. As designed, test compounds that act as pPAFAH inhibitors will prevent pPAFAH-probe interactions, thereby increasing the proportion of free (unbound) fluorescent probe in the well, leading to low fluorescence polarization. Compounds are tested in singlicate at a final nominal concentration of 3.39 μM
Protocol Summary: Prior to the start of the assay, 4.0 μL of Assay Buffer (0.01% pluronic detergent, 50 mM Tris HCl pH 8.0, 150 mM NaCl, 1 mM DTT) containing 12.5 nM pPAFAH protein were dispensed into 1536 microtiter plates. Next, 17 nL of test compound in DMSO or DMSO alone (0.34% final concentration) were added to the appropriate wells and incubated for 30 minutes at 25°C. The assay was started by dispensing 1.0 μL of 375 nM FP-Rh probe in Assay Buffer to all wells. Plates were centrifuged and incubated for 15 minutes at 25°C, and fluorescence polarization was read on a Viewlux microplate reader (PerkinElmer, Turku, Finland) using a BODIPY TMR FP filter set and a BODIPY dichroic mirror (excitation = 525 nm, emission = 598 nm) for 15 seconds for each polarization plane (parallel and perpendicular). Assay Cutoff: Compounds that inhibited pPAFAH ≥24.55% were considered active.
Confirmation uHTS assay to identify pPAFAH inhibitors (AID 463230)
Assay Overview: The purpose of this assay is to confirm activity of compounds identified as active in the primary uHTS pPAFAH screen (AID 463082). In this assay, the FP-Rh probe was used to label pPAFAH in the presence of test compounds and analyzed as described previously(AID 463082). Compounds were tested in triplicate at a nominal concentration of 3.39 μM.
Protocol Summary: The assay was performed as described previously (AID 463082), except that compounds were tested in triplicate. Assay Cutoff: Compounds that inhibited pPAFAH ≥24.55% were considered active.
Secondary Assays
Inhibition of cherry-picked uHTS hits (AID 588474)
Assay Overview: The purpose of this assay is to determine whether test compounds identified as active in the uHTS campaign (AIDs 463082 and 463230) can inhibit over-expressed pPAFAH in a complex proteomic lysate. In this assay, proteome containing pPAFAH is incubated with test compound followed by reaction with an SH-specific FP-Rh activity-based probe. The reaction products are separated by SDS-PAGE and visualized in-gel using a flatbed fluorescence scanner. The percentage activity remaining is determined by measuring the integrated optical density (IOD) of the bands. As designed, test compounds that act as pPAFAH inhibitors will prevent enzyme-probe interactions, thereby decreasing the proportion of bound fluorescent probe, giving lower fluorescence intensity in the band in the gel.
Protocol Summary: Recombinant mouse pPAFAH (25 μL of 0.25 mg/mL membrane proteome preparation from transiently transfected 293T Hek cells overexpressing pPAFAH, Open Biosystems Accession BC010726, in Dulbecco’s PBS [DPBS]) was treated with test compound (1 μL of a 25× stock in DMSO, 5 μM final concentration) or DMSO (1 μL) for 30 minutes at 37°C. FP-Rh (1 μL of 25× stock in DMSO; 2 μM final concentration) was added and the reaction was incubated for 30 minutes at 25°C, quenched with 2× SDS-PAGE loading buffer, separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the IOD of pPAFAH bands relative to a DMSO-only (no compound) control. Assay Cutoff: Compounds with ≥75% inhibition were considered active.
Inhibition and selectivity of carbamate library members (AIDs 588773 and 588774)
Assay Overview: The purpose of this assay is to determine whether test compounds (either cherry picked uHTS hits [AID 588773] or synthetic medchem compounds [AID 588774]) can inhibit over-expressed pPAFAH in a complex proteomic lysate and to estimate compound selectivity in an ABPP assay. Test compound is incubated with recombinantly-expressed target enzyme pPAFAH (Assay 1) or serine hydrolase (potential anti-target) rich complex proteome (Assay 2) followed by reaction with an SH-specific FP-Rh activity-based probe. The reaction products are separated by SDS-PAGE and analyzed as described for AID 588474.
Protocol Summary:
Assay 1: Recombinant mouse pPAFAH (50 μL of 0.25 mg/mL membrane proteome preparation from transiently transfected 293T Hek cells overexpressing pPAFAH, Open Biosystems Accession BC010726) in DPBS was treated with test compound (10, 1, or 0.1 μM final concentration, 1 μL of a 50× stock in DMSO) or DMSO (1 μL) for 30 minutes at 37°C. FP-Rh (1 μL of a 50 μM solution in DMSO; 1 μM final concentration) was added, and the reaction was incubated for 30 minutes at 37°C, quenched with an equal volume of 2× SDS-PAGE loading buffer (reducing), separated by SDS-PAGE, and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the integrated optical density of the pPAFAH band relative to a DMSO-only (no compound) control. Assay Cutoff: Compounds with ≥50% inhibition at 0.1 μM were considered active.
Assay 2: Mouse brain membrane proteome (50 μL of 1 mg/mL in DPBS) was treated with test compound (10, 1, or 0.1 μM final concentration, 1 μL of a 50× stock in DMSO) or DMSO (1 μL) for 30 minutes at 37°C. FP-Rh (1 μL of a 25 μM solution in DMSO; 1 μM final concentration) was added, and the reactions was incubated for 30 minutes at 37°C, quenched with an equal volume of 2× SDS-PAGE loading buffer (reducing), separated by SDS-PAGE, and visualized by in-gel fluorescent scanning. The percentage activity remaining for each anti-target (fatty acid amide hydrolase [FAAH], monoacylglycerol lipase [MAGL], abhydrolase domain-containing proteins 3, 4, and 6 [ABHD3, ABHD4, ABHD6], lysophospholipases 1 and 2 [LYPLA1, LYPLA2], and unidentified proteins with molecular weight [MW] of 32kDa and 30kDa) was determined by measuring the integrated optical density of the individual protein bands relative to a DMSO-only (no compound) control. Assay Cutoff: Compounds with ≥50% inhibition at 0.1 μM were considered active.
Selectivity against anti-target PAFAH2 (AID 588767)
Assay Overview: The purpose of this assay is to determine whether test compounds can inhibit the pPAFAH anti-target PAFAH2 in a gel-based competitive ABPP assay. In this assay, endogenous PAFAH2 is incubated with test compound followed by reaction with the SH-specific FP-Rh activity-based probe. The reaction products are separated by SDS-PAGE and analyzed as described for AID 588474.
Protocol Summary: Soluble proteome (1 mg/ml in DPBS) of BW5147-derived murine T cells was treated with test compound (1 μL of a 50× stock in DMSO; 0.1 μM, 1 μM, or 10 μM final concentration) or DMSO (1 μL) for 30 minutes at 37 °C. FP-Rh (1 μL of a 25 μM solution in DMSO; 1 μM final concentration) was added, and the reaction was incubated for 30 minutes at 37°C, quenched with an equal volume of 2× SDS-PAGE loading buffer (reducing), separated by SDS-PAGE, and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the IOD of the PAFAH2 band relative to a DMSO-only (no compound) control. Assay Cutoff: Compounds with ≥50% inhibition at 0.1 μM were considered active.
Determination of IC50 values against mouse pPAFAH (AIDs 588769 and 588770)
Assay Overview: The purpose of this assay is to determine the IC50 values of test compounds (either cherry picked uHTS hits [AID 588769] or synthetic medchem compounds [AID 588770]) for pPAFAH inhibition. In this assay, the SH-specific FP-Rh activity-based probe is used to label pPAFAH in the presence of test compounds. The reaction products are separated by SDS-PAGE and analyzed as described for AID 588474.
Protocol Summary: Recombinant mouse pPAFAH (50 μL of 0.25 mg/mL membrane proteome preparation from transiently transfected 293T Hek cells overexpressing pPAFAH, Open Biosystems Accession BC010726) in DPBS was treated with test compound (1 μL of a 25× stock in DMSO) or DMSO (1 μL) for 30 minutes at 37°C before the addition of FP-Rh (1 μL of 25× stock in DMSO, 1 μM final concentration). The reaction was incubated for 30 minutes at 37°C, quenched with an equal volume of 2× SDS-PAGE loading buffer (reducing), separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the IOD of the bands relative to a DMSO-only (no compound) control. IC50 values for inhibition of pPAFAH were determined from dose-response curves from three replicates at each inhibitor concentration (7-point 1:3 dilution series from 1 μM to 1 nM). Assay Cutoff: Compounds with an IC50 ≤500 nM were considered active.
Determination of IC50 values against human pPAFAH (AID 588766)
Assay Overview: The purpose of this assay is to determine the IC50 values of test compounds for human pPAFAH inhibition. In this assay, the SH-specific FP-Rh activity-based probe is used to label pPAFAH in the presence of test compounds. The reaction products are separated by SDS-PAGE and analyzed as described for AID 588474.
Protocol Summary: Recombinant, human pPAFAH (25 μL of 1 μg/mL; purified GST-fusion construct expressed in E.coli BL21 cells; protein contains residues 42–441 plus a non-native N-terminal extension (GSPNSRVD), and the following mutations in the hydrophobic patch region: I120A, L123A, L124A) in DPBS was treated with test compound (1 μL of a 25× stock in DMSO) or DMSO (1 μL) for 30 minutes at 37°C before the addition of FP-Rh (1 μL of 25× stock in DMSO, 1 μM final concentration). The reaction was incubated for 30 minutes at 37°C, quenched with an equal volume of 2× SDS-PAGE loading buffer (reducing), separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the IOD of the bands relative to a DMSO-only (no compound) control. IC50 values for inhibition of pPAFAH were determined from dose-response curves from three replicates at each inhibitor concentration (7-point 1:3 dilution series from 1 μM to 1 nM). Assay Cutoff: Compounds with an IC50 ≤500 nM were considered active.
Analysis of Cytotoxicity (AID 588768)
Assay Overview: The purpose of this assay is to determine cytotoxicity of inhibitor compounds belonging to the carbamate scaffold. In this assay, 293T Hek cells in either serum-free media (Assay 1) or media containing 10% fetal calf serum (FCS); Assay 2) are incubated with test compounds, followed by determination of cell viability. The assay utilizes the WST-1 substrate which is converted into colorimetric formazan dye by the metabolic activity of viable cells. The amount of formazan generated directly correlates to the number of metabolically active cells in the culture. As designed, compounds that reduce cell viability will result in decreased absorbance of the dye.
Protocol Summary: This assay was started by dispensing 293T Hek cells in DMEM media (100 μL, 15,000 cells/well) into a 96-well plate. Both serum-free media (Assay 1) and media supplemented with 10% FCS; Assay 2) were tested. Compound (10 μL of 11× stock solutions in media containing 10% DMSO) was added to each well, cells were incubated for 48 hours at 37°C in a humidified incubator, and cell viability was determined by the WST-1 assay (Roche) according to manufacturer instructions. CC50 values were determined from dose-response curves from five replicates at each compound concentration (7-point 1:5 dilution series starting at a nominal test concentration of 50 μM). Assay Cutoff: Compounds with a CC50 value of ≤5 μM were considered active (cytotoxic).
LC-MS/MS Analysis of Inhibitor Binding Mode (AID 588788)
Assay Overview: The purpose of this assay is to assess the covalent nature of an inhibitor compound belonging to the carbamate scaffold and determine whether or not it labels the active site serine of pPAFAH. In this assay, purified enzyme is reacted with inhibitor compound, digested with trypsin, and the resulting peptides are analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The resulting data are analyzed to identify sites of covalent labeling.
Protocol Summary: Two aliquots of purified, recombinant human pPAFAH (25 μL of 40 μM in DPBS; purified GST-fusion construct expressed in E. coli BL21 cells; protein contains residues 42–441 plus a non-native N-terminal extension [GSPNSRVD], and the following mutations in the hydrophobic patch region: I120A, L123A, L124A) were prepared. To one aliquot was added test compound (1 μL of 25× stock in DMSO, 100 μM final concentration). To the second (control) aliquot was added an equal volume of DMSO. Reactions were gently vortexed and incubated at 37°C for 60 minutes. To each reaction was added freshly prepared urea (75 μL of 6 M in DPBS), DDT (1.5 μL of 10 mM), and the reactions were incubated at 65°C for 25 minutes and then allowed to cool to room temperature. To each reaction was then added freshly prepared iodoacetamide (20 μL of 400 mM), and the reactions were rotated for 30 minutes at 25°C in the dark. Aqueous ammonium bicarbonate (150 μL of 50 mM) was added, followed by CaCl2 (1.5 μL of 100 mM), and sequencing grade modified trypsin (1 μg). Reactions were incubated at 37°C for 12 hours. The protein digests were acidified with formic acid (5% v/v final).
An Agilent 1200 series quaternary HPLC pump and Thermo Scientific LTQ-Orbitrap mass spectrometer were used for sample analysis. A fraction (20 μL) of the protein digest for each sample was pressure-loaded onto a 100 micron fused-silica column (with a 5 micron in-house pulled tip) packed with 10 cm of Aqua C18 reversed-phase packing material. Chromatography was carried out using an increasing gradient of aqueous acetonitrile containing 0.1% formic acid over 125 minutes. Data were collected in data-dependent acquisition mode with dynamic exclusion turned on (60 seconds, repeat of 1). Specifically, one full MS (MS1) scan (400–1800 m/z) was followed by 7 MS2 scans of the most abundant ions. The MS2 spectra data were extracted from the raw file using RAW Xtractor (version 1.9.1; publically available at http://fields.scripps.edu/downloads.php). The MS2 spectra generated for each run were searched against the latest version of the IPI human protein database concatenated to a reversed decoy database using Sequest [24]. Search parameters specified a static modification of +57.021 for cysteine due to alkylation, a variable modification of +253.12152 for serine to account for possible compound labeling, and no enzyme specificity. The resulting peptide identifications were assembled into protein identifications using DTASelect (version 2.0.41) using the –trypstat and --modstat options, which apply different statistical models for the analysis of tryptic, half-tryptic, non-tryptic, and modified peptides. DTASelect 2.0 uses a quadratic discriminant analysis to achieve a user-defined false positive hit rate below 1% (as determined by number of hits against the reversed database) at the peptide level. No modified peptides were identified in the DMSO-treated sample. Assay Cutoff: Compounds observed to covalently modify the active site serine of pPAFAH were considered active.
ABPP-MudPIT Analysis of Selectivity (AID 588785 and AID 588787)
Assay Overview: The purpose of this assay is to determine the selectivity profile of test compounds in a complex proteome by ABPP in combination with the multidimensional protein identification technology (MudPIT) [25, 26] LC-MS/MS analysis platform. In this assay, paired complex proteome samples are treated with test compound or DMSO, followed by reaction with the serine hydrolase specific activity-based fluorophosphonate-biotin (FP-biotin) affinity probe. Biotinylated proteins are enriched, trypsinized, and analyzed by MudPIT. Inhibition of target and anti-target activity is quantified by comparing spectra counts between the test compound- and DMSO-treated samples. As designed, compounds that act as inhibitors will block FP-biotin labeling, reducing enrichment in the inhibitor-treated sample relative to the DMSO-treated sample, thus giving a smaller spectra count ratio (test compound/DMSO) for each protein. Proteins not targeted by inhibitors would be expected to have a ratio of 1.
Protocol Summary: Sample Preparation. Paired aliquots of mouse brain soluble proteome (AID 588787) or mouse brain membrane proteome (AID 588785) (1mL of 1 mg/mL in 50 mM Tris pH 8.0 [Tris] buffer) were treated with either 1 μM test compound (20 μL of a 50× stock in DMSO) or DMSO for 30 min at 37°C. Each reaction was labeled with 5 μM of FP-biotin (5 μL of a 50× stock in DMSO) for 2 hours at 25°C. TritonX100 was added to 1% final concentration (v/v), and the proteomes were desalted over PD-10 desalting columns (GE Healthcare). SDS was added to the eluate to give 0.5% final concentration, and the samples heated to 90°C for 8 minutes. FP-labeled proteins were enriched with streptavidin beads (100 μL of 50% slurry; 25°C, 1 hour, 8.5 mL total volume Tris). Following enrichment, the beads were washed with 1% SDS in Tris (2x), 6 M Urea in Tris (2x), and Tris (2x), and then resuspended in 8 M urea, reduced with TCEP (10 mM) for 25 minutes at 25°C, and alkylated with iodoacetamide (12.5 mM) for 25 minutes at 25°C in the dark. The urea concentration was reduced to 2 M with Tris, and on-bead digestions were performed for 12 hours at 37°C with sequence-grade modified trypsin (Promega; 2 μg) in the presence of 2 mM CaCl2. Peptide samples were acidified to a final concentration of 5% (v/v) formic acid, pressure-loaded on to a biphasic (strong cation exchange/reversed phase) MudPIT capillary column and analyzed as described next. A total of 4 biological replicates were prepared and analyzed for each experiment.
LC-MS/MS analysis. Digested and acidified peptide mixtures were analyzed by two-dimensional liquid chromatography/tandem mass spectrometry (MudPIT) using an Agilent 1200-series quaternary pump and Thermo Scientific LTQ ion trap mass spectrometer. Peptides were eluted in a 5-step MudPIT experiment using 0%, 25%, 50%, 80%, and 100% salt bumps of 500 mM aqueous ammonium acetate, with chromatographic elution following each salt pulse accomplished using an increasing gradient of aqueous acetonitrile containing 0.1% formic acid over 125 minutes. Data were collected in data-dependent acquisition mode with dynamic exclusion turned on (90 s, repeat of 1). Specifically, one full MS (MS1) scan (400–1800 m/z) was followed by 7 MS2 scans of the most abundant ions. The MS2 spectra data were extracted from the raw file using RAW Xtractor (version 1.9.1; publically available at http://fields.scripps.edu/downloads.php). MS2 spectra data were searched using the PROLUCID algorithm (publically available at http://fields.scripps.edu/downloads.php) against the latest version of the mouse UniProt database concatenated with the reversed database for assessment of false-discovery rates. PROLUCID searches allowed for variable oxidation of methionine (+16), static modification of cysteine (+57 due to alkylation), and no enzyme specificity. The resulting MS2 spectra matches were assembled into protein identifications and filtered using DTASelect (version 2.0.41) using the –trypstat and –modstat options, which apply different statistical models for the analysis of tryptic, half-tryptic, non-tryptic, and modified peptides. DTASelect 2.0 uses a quadratic discriminant analysis to maintain a user-defined false positive hit rate (as determined by number of hits against the reversed database) below 1% at the peptide level. Spectra count abundance ratios (test compound/DMSO) were calculated for all serine hydrolases with an average of 10 or more spectra counts in the DMSO control samples. Assay Cutoff: A compound was considered active for a particular target/anti-target with a spectra count ratio (test compound/DMSO control) of ≤0.5.
Analysis of pPAFAH Inhibition In Situ (AID 588817)
Assay Overview: Assay Overview: The purpose of this assay is to determine whether or not test compounds can inhibit pPAFAH in situ. In this assay, cultured cells are incubated with test compound. Cells are harvested, homogenized, and reacted with the SH-specific FP-Rh activity-based probe. The reaction products are separated by SDS-PAGE and analyzed as described for AID 588474.
Protocol Summary: Transiently transfected 293T Hek cells overexpressing mouse pPAFAH (Open Biosystems Accession BC010726) were cultured in a 6-well dish for 48 hours in DMEM medium containing 10% FCS. Cells were washed with DBPS and fresh, serum-free DMEM medium (1 mL) was added. Cells were treated with test compound (10 μM, 1 μM, 0.1 μM, and 0.01 μM final concentration; 10 μL of 100× stock in DMSO) or DMSO only and incubated for 4 hours at 37°C. Cells were washed with DPBS (4x), harvested, and homogenized by sonication. The soluble and membrane fractions were separated by centrifugation (100K × g, 45 min) and the protein concentration in each fraction was adjusted to 1 mg/mL with DPBS. FP-Rh (1 μL of 50× stock in DMSO) was added to a final concentration of 1 μM in 50 μL total reaction volume. The reaction was incubated for 30 minutes at 37°C, quenched with an equal volume of 2× SDS-PAGE loading buffer, separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the integrated optical density of the pPAFAH band relative to a DMSO-only (no compound) control. Assay Cutoff: Compounds with ≥50% inhibition at 0.1 μM compound concentration were considered active.
Analysis of pPAFAH Inhibition In Vivo (AID 588789 and AID 588823)
Assay Overview: The purpose of this assay is to determine whether or not test compounds can inhibit pPAFAH in vivo (AIDs 588789 and 588823) and to assess selectivity (AID 588823). In this assay, test compounds are orally administered to mice. Mice are sacrificed, and their brain tissue harvested, homogenized, and the membrane fraction isolated and reacted with the activity-based probes HT-01 [27] or FP-Rh. HT-01 bears a BODIPY fluorophore and urea triazole reactive group that selectively labels several serine hydrolases, including pPAFAH. This reagent is used in addition to the standard SH probe FP-Rh to enhance visualization of pPAFAH, which is otherwise obscured by other SHs upon SDS-PAGE separation/visualization. The reaction products are separated by SDS-PAGE and analyzed as described for AID 588474.
Protocol Summary: Purpose-bred WT laboratory mice were orally administered test compound (50 mg/kg in PEG300 vehicle solution) or vehicle only (n = 2 or 4 per group). After four hours, mice were humanely sacrificed (anesthetized with isoflurane and decapitated) and brain tissues removed and snap frozen in liquid nitrogen. Tissues were homogenized and the membrane fraction isolated by centrifugation (45 min, 100K × g) and adjusted to 1 mg/mL in DPBS. Aliquots (50 μL) were treated with HT-01 (1 μL of 50× stock in DMSO, 1 μM final concentration) or FP-Rh (1 μL of 50× stock in DMSO, 1 μM final concentration). The reaction was incubated for 30 minutes at 37°C, quenched with an equal volume of 2× SDS-PAGE loading buffer (reducing), separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the IOD of test compound bands relative to vehicle bands. Assay Cutoff: Compounds with ≥50% inhibition were considered active.
2.2. Probe Chemical Characterization

CID 53364507
SID 125269079
ML256
The probe structure was verified by 1H NMR (see section 2.3) and high resolution MS (m/z calculated for C26H24N4O2 [M+H]+: 425.1972, found 425.1974). Purity was assessed to be 96% by NMR. Solubility (room temperature) was determined to be 16.3 μM in PBS, 7.5 μM in DMEM medium, and >100 μM in DMEM medium containing 10% fetal calf serum. Stability in PBS was determined by LC-MS to be >48 hours.
Table 2.2-1Compounds submitted to the SMR collection
| Designation | CID | SID | SRID | MLS |
|---|---|---|---|---|
| Probe | 53364507 | 125269079 | SR-02000001715-1 | MLS003874819 |
| Analog 1 | 46829224 | 99205832 | SR-02000000421-1 | MLS003874820 |
| Analog 2 | 53364494 | 125269060 | SR-02000001696-1 | MLS003874821 |
| Analog 3 | 53364529 | 125269074 | SR-02000001710-1 | MLS003874822 |
| Analog 4 | 53364548 | 125269075 | SR-02000001711-1 | MLS003874823 |
| Analog 5 | 53364487 | 125269077 | SR-02000001713-1 | MLS003874824 |
2.3. Probe Preparation

2-methoxy-5-phenylpyridine: A mixture of 5-bromo-2-methoxypyridine (5.641 g, 30 mmol, 1.0 eq), phenylboronic acid (4.389 g, 36 mmol, 1.2 eq), Na2CO3 (9.539 g, 90 mmol, 3.0 eq) and Pd(Ph3P)4 (1.74 g, 5 mmol%) in toluene/H2O (v/v 5:3, 240 mL) was heated to 90°C under N2 overnight. The product was extracted with EtOAc (3 × 300 mL) and purified by silica gel column chromatography (1–10% ethyl acetate/hexanes) to afford 2-methoxy-5-phenylpyridine (5.0571g, 27 mmol, 90%).
5-phenylpyridin-2-ol: To a solution of a 2-methoxy-5-phenylpyridine (3.13 g, 17 mmol, 1.0 eq) in dry CH2Cl2 (85 mL) was slowly added BBr3 (5 g, 20 mmol, 1.2 eq) at −20°C. Upon warming to room temperature, the reaction mixture was stirred overnight. 1N HCl (34 mL) was added to quench the reaction at 0°C. The product was extracted with ethyl acetate (3 × 170 mL). The combined organic phases were dried over NaSO4, filtered and concentrated under reduced pressure. The product was purified by silica gel column chromatography (1–10% methanol/CH2Cl2) to recover unreacted starting material and afford 5-phenylpyridin-2-ol (0.0825 g, 0.5 mmol, 3%).
5-phenylpyridin-2-yl 4-(2-methylquinolin-4-yl)piperazine-1-carboxylate (Probe ML256): To a solution of triphosgene (0.0549 g, 0.185 mmol, 0.33 eq) in CH2Cl2 (5.55 mL), was slowly added 5-phenylpyridin-2-ol (0.0951 g, 0.555 mmol, 1.00 eq) and DIEA (97 μL, 0.0716 g, 0.555 mmol, 1.00 eq) as a solution in THF (5.55 mL). The reaction was stirred at room temperature for 30 min to generate the intermediate chloroformate. In a separate flask containing CH2Cl2 (5.55 mL), 2-methyl-4-piperazinoquinoline (0.1892 g, 0.833 mmol, 1.50 eq) was mixed with DIEA (146 μL, 0.1078 g, 0.833 mmol, 1.5 eq). The mixture was stirred for 5 minutes at room temperature and then dropped into the flask containing the chloroformate. The reaction mixture was stirred at room temperature for another 30 minutes and partitioned between CH2Cl2 (28 mL) and saturated NH4Cl (28 mL). The organic layer was washed one more time with saturated NH4Cl (17 mL), and dried over MgSO4. The solvent was removed by rotary evaporation and the product was purified by silica gel column chromatography (1:1 ethyl acetate/hexanes) to afford 5-phenylpyridin-2-yl 4-(2-methylquinolin-4-yl)piperazine-1-carboxylate (Probe ML256) (0.0903 g, 0.212 mmol, 38%). 1H NMR (400 MHz, CDCl3) δ 8.59 (dd, J = 2.5, 0.7 Hz, 1H), 8.04–7.91 (m, 3H), 7.65 (ddd, J = 8.3, 6.8, 1.4 Hz, 1H), 7.60–7.35 (m, 6H), 7.22 (dd, J = 8.4, 0.7 Hz, 1H), 6.78 (s, 1H), 4.02 (t, J = 4.9 Hz, 2H), 3.90 (t, J = 4.8 Hz, 2H), 3.2 (t, J = 5.2 Hz, 4H), 2.70 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 159.71, 157.84, 156.62, 153.10, 149.50, 146.76, 138.30, 137.19, 135.33, 129.56, 129.50, 129.32, 128.35, 127.34, 125.17, 123.20, 121.95, 116.31, 110.07, 52.20, 52.08, 45.08, 44.30, 25.87; HRMS ESI-TOF high-acc (M + H+) calculated for C26H24N4O2 425.1972, found 425.1974; purity estimate by NMR: 96%.
3. Results
Probe ML256 has IC50 values of 31 nM and 6 nM against the mouse and human isoforms of pPAFAH, respectively (Section 3.2). ML256 also exhibits high selectivity (>322-fold) vs. more than 20 brain SHs and its closest homolog, PAFAH2, as assayed by gel-based ABPP (Sections 3.4 and 3.6). Selectivity was also confirmed for the more distantly related PAFAH family members PAFAH1b2 and PAFAH1b3 and 40+ SH enzymes by ABPP-MudPIT (Section 3.6). ML256 was shown to be highly active against pPAFAH both in situ and in vivo (Section 3.5).
3.1. Summary of Screening Results
In the primary fluopol-ABPP pPAFAH HTS Assay (AID 463082), ~326K compounds were tested for inhibition of enzyme labeling by the SH-specific activity-based probe FP-Rh [18]. The assay was conducted in singlicate at 3.39 μM compound concentration using purified, recombinant human enzyme. A total of 4,934 compounds (1.5%) were active, passing the set threshold (mean + 3× standard deviation) of 24.55% inhibition. A total of 2,500 compounds were selected for testing in the confirmation assay (AID 463230), in which compounds were re-tested at the same concentration in triplicate. Of the 2,341 available compounds, 1,675 (71.6%) confirmed as active (Figure 3.1-1).

Figure 3.1-1
Flow chart describing HTS results.
Because the fluopol-ABPP HTS assay was conducted with purified, recombinant enzyme, we selected a subset of active compounds for secondary gel-based screening to assess pPAFAH inhibition in a complex proteome. Compounds were cherry-picked from among the top HTS hits using the following criteria: in the confirmation HTS assay, a total of 407 compounds inhibited the target enzyme by at least 60%. Of those 407 compounds, 87 were removed due to reactivity with other select enzymes screened by fluopol-ABPP: GST01 (68, AID 1974), PME1 (5, AID 2130), PREPL (13, AID 2751), and PAD4 (1, AID 485272). Of the remaining 320 compounds, 92 exhibited more than 5% activity in all bioassays tested and were removed. Out of this group of 228 compounds, just over half were cherry picked for secondary screening based on lack of undesired functional groups (esters, hydrazines, oximes, etc.) and medchem potential. Because the carbamate scaffold was observed to feature prominently among the top HTS hits, an additional 25 carbamates were also selected for a total of 153 compounds.
For the gel-based competitive ABPP assay, a complex membrane proteome lysate prepared from 293T Hek cells overexpressing recombinant mouse pPAFAH was incubated with test compound (5 μM) followed by reaction with the FP-Rh activity-based probe. The reaction products were separated by SDS-PAGE and visualized in-gel using a flatbed fluorescence scanner. Test compounds that act as pPAFAH inhibitors prevent enzyme-probe interactions, thereby decreasing the fluorescence intensity of the protein bands. As reported in AID 588474, only twelve compounds (Table 3.1-1) inhibited the target enzyme by ≥75%, eight of which were carbamates (See Figure S1 and Table S1 for gel images and full tabulated results, respectively). Most of the carbamate compounds showed good selectivity vs. PAFAH2, the closest sequence homolog of pPAFAH, in the counterscreen HTS fluopol-ABPP assay, indicating that medchem optimization should be able to deliver a selective pPAFAH inhibitor.
Table 3.1-1
Most promising pPAFAH inhibitor leads by gel-based competitive ABPP.
We were excited to discover promising carbamate leads for pPAFAH, as we and others have had previous success developing potent and selective serine hydrolase inhibitors based on the carbamate scaffold [28–33] (Table 3.1-2). The carbamate scaffold readily lends itself to diversity-oriented synthesis and medchem manipulation. Moreover, as a class, carbamates are typically active in vivo and have the ability to penetrate the nervous system [28, 33, 34]. We have confirmed by click chemistry-ABPP that members of this chemotype show negligible reactivity with proteins outside the SH class [35]. Within the SH class, we have previously identified carboxylesterases (CESs) as common anti-targets for carbamate inhibitors [28, 35]. In this regard, the carbamate is not unique, as the CES enzymes appear to be common targets for a variety of SH-directed inhibitors [36], likely a reflection of the role they play in xenobiotic metabolism. However, CESs are mostly restricted in their expression profile to liver [37], thus their presence as anti-targets should not affect SH characterization in most cells and tissues. For example, both the FAAH inhibitor URB597 [31] and the MAGL inhibitor JZL184 [33] (see structures Table 3.1-2) have proven to be highly useful chemical probes [31, 33, 38–40] despite having some CES inhibitory activity [35, 41]. Unless the target of interest is a liver enzyme, the key selectivity requirement for successful carbamate probes is to show high selectivity among all non-CES SHs. In the case of pPAFAH, we are interested in studying the effects of enzyme inhibition in the brain, so CES inhibitory activity should not be a significant concern. We have also previously addressed the issue of imperfectly selective probes by generating control “anti-probes” (see ML211 and ML226 Probe Reports), which are structurally and mechanistically similar to the optimized inhibitor but lack activity against the SH of interest, for use in parallel biological experiments to confirm functional assignments.
Table 3.1-2
Representative carbamate SH inhibitors.
Also included in Table 3.1-2 is a carbamate lead inhibitor for pPAFAH, WWL153, which we identified from a small, in-house carbamate screening library [28]. The compound exhibited decent potency for the target enzyme, but poor selectivity, inhibiting anti-targets fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) with only a marginal dosing window (Table 3.4-1). Fortuitously, the HTS carbamate hits gave us new medchem leads to further develop this scaffold (detailed in Section 3.4) ultimately leading us to the optimized pPAFAH probe ML256 (SID 125269079).
Table 3.4-1
Target SAR Analysis.
3.2. Dose Response Curves for Probe
IC50 values for both the human (6 nM; AID 588766) and mouse (31 nM; AID 588770) isoforms of pPAFAH were obtained from gel-based competitive-ABPP data (Figure 3.2-1).
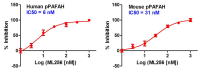
Figure 3.2-1
IC50 curves for probe ML256 (SID 125269079) as determined by gel-based competitive-ABPP with FP-Rh against purified, recombinant human pPAFAH (left) and overexpressed mouse pPAFAH in a complex proteome lysate (293T Hek membrane) (right).
3.3. Scaffold/Moiety Chemical Liabilities
The probe compound was determined to covalently modify the catalytic serine (Ser273) of pPAFAH by LC-MS/MS analysis (AID 588788). The observed mass shift of the active site peptide corresponds to carbamoylation of the enzyme upon serine nucleophilic attack at the carbonyl followed by loss of the hydroxyl moiety (Figure 3.3-1), as expected of this chemotype [35].

Figure 3.3-1
Covalent modification of pPAFAH by ML256 (SID 125269079) as determined by LC-MS/MS analysis.
The probe compound showed no reactivity with glutathione (100 μM), indicating that it has a tempered electrophilicity and specific structural elements that direct reactivity towards pPAFAH. Gel-based and LC-MS/MS (MudPIT)-based ABPP selectivity profiling also confirmed high target specificity (Section 3.6).
We [42], and others [43–45], have found that irreversible inhibitors such as ML256 offer many advantages over reversible inhibitors as pharmacologic probes for enzyme characterization in biological systems. Perhaps most importantly, it is technically straightforward to confirm the irreversible inhibition of enzymes in living systems (including mice) using competitive ABPP and click chemistry ABPP; as such, we can define the precise quantity of inhibitor and treatment time required to selectively inactivate an enzyme of interest for biological studies. This information is extremely valuable for guiding the functional characterization of SHs. Additionally, required dosing is often lower, irreversible compounds are not as sensitive to pharmacokinetic parameters, and administration can induce long-lasting inhibition [42]. In the case of the EGFR inhibitor PD 0169414, its irreversibility and high selectivity were credited with producing prolonged inhibition of the target, alleviating concerns over short plasma half-lives and reducing the need for high peak plasma levels, thus minimizing potential nonspecific toxic effects [46].
Indeed, over a third of enzymatic drug targets are irreversibly inhibited by currently marketed drugs [47]. Examples of covalent enzyme-inhibitor pairs include serine type D-Ala-D-Ala carboxypeptidase, which is covalently modified by all B-lactam antibiotics, acetylcholinesterase, whose active site serine undergoes covalent modification by pyridostigmine, prostaglandin-endoperoxide synthase, which is the target of the ubiquitously prescribed aspirin, aromatase, which is irreversibly modified by exemestane, monoamine oxidase, which is covalently modified by L-deprenyl, thymidylate synthase, which is covalently modified by floxuridine, H+/K+ ATPase, which undergoes covalent modification by omeprazole, esomeprazole, and lansoprazole, and triacylglycerol lipase, whose serine nucleophile is targeted by orlistat [47].
3.4. SAR Tables
The SAR Table 3.4-1 includes 21 compounds: the top 8 carbamates identified by HTS (entries 1–8), our in-house carbamate lead WWL153 (entry 9), and 12 new synthetic compounds (entries 10–21). The analogs are variable at the left hand binding group and right hand leaving group substituents. To investigate whether or not the carbamate was the best electrophile for pPAFAH inhibition, we also generated a series of five triazole urea analogs (Table 3.4-2). The triazole urea reactive group is another “privileged” scaffold for SH inhibition from which we have derived several potent, selective, and in situ-active SH probes: ML211 as a dual inhibitor of LYPLA1&2, ML226 for ABHD11, and ML225 for the pPAFAH homolog PAFAH2 (see also ref. [48]).
Table 3.4-2
Target SAR Analysis: Triazole Ureas.
All SAR compounds were subject to gel-based competitive ABPP profiling [19] to assess both potency and selectivity against several dozen FP-sensitive SHs. Because endogenous pPAFAH is difficult to visualize by gel due to low abundance and/or overlapping SHs, potency was assessed against recombinant mouse pPAFAH in the membrane proteome of 293T Hek cells engineered to overexpress the enzyme. Given our interest in exploring pPAFAH biochemistry in the brain and owing to the brain’s rich diversity of anti-target SHs, selectivity was assessed using a mouse brain membrane proteome preparation. For both the potency and selectivity analysis, compounds were tested at three concentrations: 10 μM, 1 μM, and 100 nM, and the gel profiling results are included in Section 3.6 (see Section 2.1 and AIDs 588773 and 588774 for protocol details). Proteins are listed as anti-targets for a given compound concentration if at least 50% inhibition was observed relative to the DMSO only (no compound) control. For comparison, we included the four non-carbamate HTS hits from Table 3.1-1 in our investigation of potency and selectivity, the results (none showed pronounced inhibition of the target enzyme) are tabulated in Table S2.
J11/WWL153 Analogs: The development of a pPAFAH inhibitor began by combining structural elements of the top HTS hits, chiefly J11 (SID 26727296, entry 1) and P09 (SID 4257433, entry 2), with our in-house lead WWL153 (SID 99205832, entry 9) [28]. WWL153 displays an IC50 on the order of ~200 nM towards pPAFAH with some inhibition of FAAH and MAGL at higher (1 μM and 10 μM) concentrations, which we wanted to avoid with the final compound. We hypothesized that the 2-methyl-4-piperazinoquinoline motif was providing the majority of the potency and selectivity for our compound, especially given the predominance of piperidine-like motifs in the structures of the 12 HTS hit compounds (all but J11, Table 3.1-1). In contrast, the leaving group moieties of the HTS carbamates, while all aromatic with an emphasis on bis/bi aromatic systems (e.g., 4-hydroxybiphenyl for P09 and P03, 4-phenoxyphenol for A07; Table 3.4-1, entries 2, 6, and 7, respectively) evinced considerably more diversity. An especially intriguing motif was the 6-chlorobenzo[d]isoxazol-3-ol leaving group of compound J11 (entry 1). Out of all the HTS carbamates, J11 displayed the best inhibition of pPAFAH in gel-based analysis (Tables 3.1-1 and 3.4-1). However, it also showed a strong potency towards the anti-targets FAAH and ABHD6, with persistent inhibition of the former even at low (100 nM) concentration (Table 3.4-1). We hypothesized that this lack in selectivity could be coming from the very simple dimethyl-amino binding group and that a more complex amine may provide better selectivity.
Based on this analysis, we synthesized a series of first generation analogs by coupling the 6-chlorobenzo[d]isoxazol-3-ol leaving group of J11 with the 2-methyl-4-piperazinoquinoline binding group of WWL153 to create JMN5, and with two prevalent amines among the HTS hits: piperidine to generate JMN6, and morpholine to give JMN10 (Table 3.4-1, entries 10–12). During synthesis, it was found that urea structural isomers of the 6-chlorobenzo[d]isoxazol-3-ol carbamates were formed as minor byproducts of the reaction; thus JMN7 (isomer of JMN5) and JMN11 (isomer of JMN10) were also synthesized and isolated for testing (Table 3.4-1, entries 13 and 14). As a group, these 6-chlorobenzo[d]isoxazol-3-ol carbamates and ureas (entries 10–14), show high potency for pPAFAH (>80% inhibition of pPAFAH at 0.1 uM; Table 3.4-1) but they also inhibited numerous other SHs, including FAAH, MAGL, and ABHD6, with ~equal potency. This anti-target activity suggests that the 6-chlorobenzo[d]isoxazol-3-ol leaving group overly activates the carbonyl, and that less activating leaving groups may provide better selectivity.
WWL153/Triazole Urea Analogs: Recently, our lab has investigated 1,2,3-triazole ureas as SH inhibitors (See ref. [48] and ML211, ML225 and ML226 Probe Reports). Given the potency of the urea JMN7 (entry 13) and the fact that we have generated selective triazole urea SH inhibitors in the past, we thought investigation of the triazole urea electrophilic scaffold for pPAFAH inhibition was merited. As such, we synthesized a series of triazole urea analogs using the 2-methyl-4-piperazinoquinoline binding group in combination with and a series of different biaromatic triazole leaving groups (Table 3.4-2). These compounds showed great potency towards pPAFAH (near complete inhibition observed at 0.1 μM compound concentration for all); however, they also inhibited FAAH, MAGL, and numerous other SH anti-targets with high potency as well. Due to the lack of selectivity, the triazole urea was deprioritized as scaffold for generation of a pPAFAH inhibitor.
P09/WWL153 Analogs: As mentioned before, one leaving group motif that was prevalent among the HTS carbamate hits was the 4-hydroxybiphenyl (P09 and P03, Table 3.4-1, entries 2 and 6). These compounds were intriguing because they not only have decent potency for inhibiting pPAFAH, but also avoid inhibiting FAAH at low concentration. The 4-hydroxybiphenyl leaving group was thus combined with the 2-methyl-4-piperazinoquinoline binding group to form JMN4 (Table 3.4-1, entry 15), which showed excellent potency for pPAFAH (IC50 = 91) and the highest selectivity coefficient (>109-fold) of any synthetic compounds tested thus far.
Since the 4-hydroxybiphenyl leaving group seemed important for avoiding FAAH inhibition, presumably due to its steric bulk, we proceeded to synthesize several JMN4 analogs with biaryl or aryl-cyclo leaving groups (JMN18-JMN23, Table 3.4-1, entries 16–21). With the exception of JMN21 (entry 19), all analogs displayed reduced potency (and reduced selectivity in the case of JMN20, entry 18) in comparison to JMN4. Compound JMN21 was ~3× more potent (IC50 = 31 nM) than JMN4 and also exhibited no anti-target effects at the highest concentration (10 μM) tested. The only difference between JMN21 and JMN4 is the replacement of a carbon in the phenol group with a nitrogen atom. This modification slightly activates the carbonyl, thus increasing potency. However, the biaryl system of JMN21 prevents promiscuous binding, thus restricting this heightened reactivity to the target enzyme. This analog, JMN21, was designated as pPAFAH probe ML256.
Summary: As compared to our initial HTS (J11 and P09) and in-house (WWL153) lead pPAFAH inhibitors, probe ML256 (IC50 31 nM) represents a 5-fold improvement in potency and two orders of magnitude improvement in selectivity, showing no evidence of FP-sensitive SH anti-target reactivity up to 10 μM compound concentration by gel-based ABPP.
3.5. Cellular Activity
In Situ Inhibition: Probe ML256 (SID 125269079) is active against pPAFAH in situ (AID 588817), inhibiting enzymatic activity by 58% at 100 nM compound concentration after 4 hours as assayed by gel-based competitive ABPP with FP-Rh (Figure 3.5-1). This result indicates that ML256 is free to cross cell membranes and inhibit its target in cells.

Figure 3.5-1
Inhibition of pPAFAH activity in situ by probe ML256 (SID 125269079). Transiently transfected 293T Hek cells overexpressing mouse pPAFAH (serum-free DMEM medium) were treated with ML256 for 4 hours, washed, harvested and the membrane fraction was isolated (more...)
In Vivo Inhibition: We also assessed the ability of probe ML256 (SID 125269079) to inhibit pPAFAH in vivo (AID 588823). Brain tissue was harvested from mice administered ML256 (50 mg/kg, oral gavage, 4 hour timecourse) and the membrane fraction isolated and profiled by gel-based competitive ABPP using either HT-01 (Figure 3.5-2, left panel) or FP-Rh (Figure 3.5-2, right panel). HT-01 is a triazole urea activity-based probe [27] that labels a handful of SHs in the brain, allowing enhanced visualization of pPAFAH, which is otherwise obscured by overlapping SH bands with FP-Rh profiling. In probe-treated animals, pPAFAH activity is reduced by more than 95%.
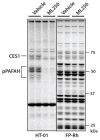
Figure 3.5-2
Inhibition of pPAFAH activity in vivo by probe ML256 (SID 125269079). Mice (n=2 per group) were administered ML256 (50 mg/kg via oral gavage) or vehicle only. After 4 hours, mice were sacrificed and their brain tissue removed. The membrane fraction was (more...)
There is one observed CES anti-target, CES1, which is inhibited by ML256 under these conditions (Figure 3.5-2A, left panel); however, CES1 is a liver enzyme [28, 37], and its presence in the membrane proteome is an artifact of blood contamination. We therefore do not believe that ML256’s anti-target activity against CES1 inhibition will unduly affect biochemical profiling of pPAFAH in the brain. Regardless, we have identified other carbamate inhibitors that also inhibit CES1, but do not affect pPAFAH (e.g., WWL47, Table 3.1-1 [28], or the in vivo-active dual KIAA1363/CES1 inhibitor JW480 [29]), and therefore could be used as “anti-probes” for biological studies. We have previously employed a similar anti-probe strategy for the LYPLA1/2 dual inhibitor ML211, which showed inhibitory activity against anti-target ABHD11, for which we developed a selective, control “anti-probe”, ML226. Other than CES1, no other anti-targets in the brain membrane proteome are observed (Figure 3.5-2A, right panel), a clean selectivity profile that is further substantiated by more in depth LC-MS/MS based profiling as detailed in Section 3.6.
Demonstrated oral bioavailability, penetration of the blood-brain barrier to deliver near complete inhibition, and a largely clean selectivity profile strongly support the utility of this probe for in vivo functional and biochemical characterization of pPAFAH. It should be noted that the irreversible nature of ML256 greatly facilitates these kinds of detailed potency and selectivity analysis in living systems.
Cytotoxicity: The probe ML256 (SID 125269079) and analogs JMN4 (SID 125269060) and WWL153 (SID 99205832) were evaluated for cytotoxicity (AID 588768) in 293T Hek cells cultured in both serum-free and serum-supplemented medium. As shown in Figure 3.5-3, ML256 had significantly higher CC50 values than either analog, affording a large dosing window (50-fold) about the in situ IC50 value of <100 nM (Figure 3.5-1).

Figure 3.5-3
Cytotoxicity anaysis of probe ML256 and analogs JMN4 and WWL153 in serum-free (A) and serum-supplemented (B) medium (AID 588768). 293T Hek cells cultured in DMEM medium with and without 10% FCS were treated with test compound and viability was assessed (more...)
3.6. Profiling Assays
HTS Carbamates: To date, the HTS hit carbamates (Table 3.1-1) have all been tested in several hundred other cell-based and non-cell based bioassays deposited in PubChem, and have shown an average hit rate of 2.1%. The most reactive carbamate, A06, with a hit rate of 3.5%, also has a Michael acceptor functionality, which could explain its increased reactivity. This low hit rate indicates that this compound class may not be generally active. No HTS activity data are yet available for probe ML256 or any of the synthetic analogs, nor has ML256 been submitted for commercial or non-commercial broad panel screening.
Gel-based Competitive ABPP: Probe ML256 (SID 125269079) and analogs listed in Tables 3.4-1 and 3.4-2 have been subject to gel-based competitive ABPP screening to assess SH reactivity against more than 20 FP-sensitive SHs (including lipases, esterases, proteases, and uncharacterized hydrolases) visible by 1D SDS-PAGE separation and fluorescent detection. This medium-throughput proteome-wide screening technique was instrumental in our medchem optimization of the probe compound (Section 3.4), allowing rapid assessment of potency and selectivity, as visualized by disappearance of bands in compound-treated lanes relative to the DMSO-only control.
For comparative assessment of compound potency (AIDs 588773 [HTS hits] and 588774 [synthetic compounds]), Figure 3.6-1 shows inhibition of recombinantly-expressed mouse pPAFAH in membrane proteome preparation from transiently transfected 293T Hek cells overexpressing the enzyme at three compound concentrations (10 μM, 1 μM, and 0.1 μM). All data are summarized in Tables 3.4-1, 3.4-2, and S2. As a class, the triazole ureas were the most potent compounds (showing most complete inhibition at 0.1 μM compound concentration), but, as discussed in Section 3.4, they exhibited poor selectivity profiles (see Figure 3.6-3, lower left panel).

Figure 3.6-1
Potency of ML256 (SID 125269079) and analogs against recombinant mouse pPAFAH in a complex proteome (293T Hek cell membranes overexpressing the enzyme). For this gel-based competitive ABPP assay, proteome was incubated with test compound (10 μM, (more...)

Figure 3.6-3
Selectivity of synthetic SAR compounds as assessed by gel-based competitive ABPP in the mouse brain membrane proteome. See Tables 3.4-1 and 3.4-2 for summary of selectivity results. pPAFAH is not visible in these gels due to overlapping SH bands. See (more...)
For selectivity analysis, the competitive ABPP experiment was repeated in mouse brain membrane proteome (AIDs 588773 [HTS hits] and 588774 [synthetic compounds]). This proteome was chosen due to its relevance for future study of pPAFAH in the nervous system as well as its diversity of SHs. pPAFAH is not visible due to overlapping SH bands. Compounds were tested at the same three concentrations (10 μM, 1 μM, and 0.1 μM) for determination of fold selectivity as reported in the tables in Section 3.4. Proteins are listed as anti-targets (Tables Section 3.4 and S2) if at least 50% inhibition is observed (as quantified relative to the DMSO control) for a given compound concentration. HTS hits are shown in Figure 3.6-2 and synthetic analogs are shown in Figure 3.6-3. Overall, this gel-based competitive ABPP analysis revealed that ML256 (Figure 3.6-3, top left panel) shows no anti-target inhibition at 10 μM, corresponding to a large (>322-fold) selectivity window over all (>20) FP-sensitive SHs resolvable by 1D SDS-PAGE.

Figure 3.6-2
Selectivity of HTS hit compounds as assessed by gel-based competitive ABPP with FP-Rh in the mouse brain membrane proteome. See Tables 3.4-1 and S2 for summary of selectivity results. pPAFAH is not visible in these gels due to overlapping SH bands. See (more...)
Selectivity vs. PAFAH2: Because PAFAH2 is the closest sequence homolog to pPAFAH, we wanted to explicitly test the reactivity of ML256 (SID 125269079) towards this enzyme by gel-based competitive ABPP (AID 588767). For this assay, the soluble proteome of BW5147-derived murine T cells, which endogenously express PAFAH2, was treated with ML256 (10 μM, 1 μM, and 0.1 μM) or DMSO for 30 minutes followed by reaction with FP-Rh. Treatment with AA39-5 (SID 103913575), a compound known to inhibit PAFAH2 (see ML225 Probe Report), is shown as a control. No inhibition of PAFAH2 is observed at 10 μM ML256 concentration, giving fold selectivity of >322 for pPAFAH vs. PAFAH2.

Figure 3.6-4Selectivity of ML256 (SID 125269079) vs. PAFAH2, the closest sequence homolog of pPAFAH, as assessed by gel-based competitive ABPP with FP-Rh in the murine T-cell soluble proteome
No inhibition of anti-target PAFAH2 is observed at the highest concentration (10 μM) tested relative to the DMSO control, giving a fold selectivity of >322. Known PAFAH2 inhibitor AA39-5 (SID 103913575) is shown as a control. Fluorescent images shown in grey scale.
ABPP-MudPIT: To more comprehensively identify potential anti-targets, we utilized a quantitative LC-MS/MS-based platform termed competitive ABPP-MudPIT (Figure 3.6-5A) [49]. ABPP-MudPIT allows for robust quantitation of inhibited enzymes via comparison of the spectra counts peptides from control vs. inhibitor-treated samples. Paired samples of mouse brain membrane (AID 588785) and soluble (AID 588787) proteomes were treated with 1 μM of ML256 or DMSO (30 minutes) followed by reaction with the affinity-tagged FP-biotin activity-based probe (5 μM, 2 hours). Reactions were enriched with streptavidin, digested on-bead with trypsin, and analyzed by MudPIT [25, 26] using an LTQ Thermo Scientific instrument. Protein identifications and spectra counts were obtained by MS2 searching using the Prolucid (http://fields.scripps.edu/researchtools.php) search algorithm and filtered using DTASelect [50]. The depicted bar graphs show the relative spectra counts between inhibitor- and DMSO-treated samples for each SH identified. Enzymes susceptible to inhibition upon compound treatment would be expected to have significantly reduced spectra counts in the inhibitor-treated sample. The results (Figure 3.6-5B and C) demonstrate that ML256 exhibits significant selectivity for pPAFAH, blocking 100% of activity while not affecting any of the other 40+ SHs detected in the mouse brain proteome, with the exception of the liver enzyme CES1, consistent with gel-based profiling results (see Section 3.5). The other PAFAH family enzymes PAFAH1b2 and PAFAH1b3 also showed no evidence of ML256 inhibition.

Figure 3.6-5
Potency and selectivity of ML256 assessed by ABPP-MudPIT. A) Overview of the ABPP-MudPIT method. B and C) Inhibition profiles of ML256 (1 μM, 2 hour) in the mouse brain membrane (B) and soluble (C) fractions shows significant inhibition of only (more...)
Of note, we have previously determined that we could detect the SH fatty acid amide hydrolase (FAAH) at concentrations as low as 0.0005% of the total proteome (~200 copies per cell) by gel-based ABPP [18]. By comparison, we estimate that the sensitivity of a standard ABPP-MudPIT assay is at least 10-fold higher (0.00005% of the total cell proteome, or 20 copies per cell). As such, both gel-based and LC-MS/MS-based methods offer detection of low abundance SHs for a comprehensive selectivity analysis.
4. Discussion
Probe ML256 (SID 125269079) was identified as a highly potent and selective covalent inhibitor of the target enzyme pPAFAH. The probe has an IC50 of 31 nM against the mouse isoform and 6 nM against the human isoform of pPAFAH (Section 3.2), and very high selectivity (>322-fold) against its closest homologue PAFAH2 and all other brain-specific SH anti-targets surveyed by gel-based competitive ABPP (Sections 3.4 and 3.6). A more in-depth ABPP-MudPIT analysis revealed no anti-target reactivity for more than 40 SHs, with the exception of the liver enzyme CES1 (Section 3.6). While this anti-target activity is not expected to be problematic for investigation of the brain biology of pPAFAH, there are several CES1 inhibitors available for use as controls in biological experiments [28, 29]. ML256 is active in situ with an IC50 <100 nM, orally bioavailable, active in vivo, and able to cross the blood-brain to inhibit its intended target to near completion in the brain (Section 3.5). Importantly, the compound shows no evidence of cytotoxicity up to 6 μM (Section 3.5), is stable in PBS for more than 48 hours, and easily synthesized via a three-step protocol (Section 2.2). Taken together, these findings suggest that it is very possible to develop potent and selective probes based on tempered electrophilic scaffolds, like the carbamate, and that ML256 will be a highly successful probe for investigation of pPAFAH biology and active-site architecture.
4.1. Comparison to existing art and how the new probe is an improvement
There were two existing pPAFAH inhibitors when we initiated our HTS campaign for inhibitor discovery: the carbamate WWL153 (Table 3.4-1, entry 9) and darapladib, a highly potent (IC50 270 pM) reversible inhibitor of pPAFAH (Figure 4.1-1) currently in phase III clinical trials for treatment of atherosclerosis [21, 51–55].

Figure 4.1-1
Structure of darapladib, a GlaxoSmithKline drug in phase III clinical trials for the treatment of atherosclerosis.
WWL153 was our best lead inhibitor for pPAFAH inhibition, and ML256 is a significant improvement in potency (>5-fold), selectivity (two orders of magnitude), and cytotoxic properties (>100-fold). We have also demonstrated that ML256 is active both in situ and in vivo. The inhibitory properties of WWL153 in living systems have not been investigated.
Darapladib is a highly potent inhibitor of pPAFAH that operates by a reversible (non-covalent) mechanism, in contrast to the irreversible, covalent mechanism of ML256. As demonstrated in Section 3.5, a key advantage of ML256 is that its irreversible mechanism of action enables, when combined with ABPP methods, direct confirmation of pPAFAH inhibition in vivo. Thus, persistent questions, such as the endogenous substrates of pPAFAH and the enzyme’s function in the mammalian nervous system, can be assessed in mouse models with ML256. Such studies are more difficult to perform with a reversible inhibitor like darapladib, given that there are no known biomarkers that can be used to confirm the reversible inhibition of pPAFAH in living systems. As such, we view the irreversible probe ML256 as a critical addition to the chemical tools available for manipulation of pPAFAH activity for biochemical investigation.
Additionally, while darapladib is not commercially available (and very difficult to obtain from GlaxoSmithKline in sufficient quantities for comprehensive biological studies), we will make ML256 available to any investigator interested in studying pPAFAH biology. Unfortunately there is also no straightforward way to circumvent darapladib commercial unavailability through in-house synthesis. There is no detailed synthetic route published for the compound, and the synthetic overview provided in Bioorg. Med. Chem. Lett. [21] was not sufficiently detailed to allow, in our hands, successful resynthesis of the compound. In contrast, ML256 is easily synthesized in three steps from commercially available starting materials and a detailed protocol is included in Section 2.3.
Finally, one key aim of our HTS inhibitor discovery campaign was to find new scaffolds for co-crystallization studies of pPAFAH-inhibitor complexes to further define the active site and substrate specificity of pPAFAH. Attempts to crystallize pPAFAH with darapladib have been unsuccessful; however, the medchem campaign to optimize a lead carbamate inhibitor for pPAFAH has generated to a diversity of potent analogs that may lend themselves to crystallographic study (in progress).
4.2. Mechanism of Action Studies
As determined from LC-MS/MS analysis (AID 588788), the probe is an activity-based inhibitor that covalently labels the active site serine nucleophile, Ser273, of pPAFAH (section 3.3). The observed mass shift of the active site peptide is consistent with carbamoylation of the enzyme (Figure 3.3-1).
4.3. Planned Future Studies
We plan to comprehensively establish parameters for in situ and in vivo use and explore the target specificity of ML256 with application of alkyne analogs via click chemistry ABPP. For biological application, we plan to use ML256 as a probe for investigation of the potential role of pPAFAH in inflammatory processes by global proteomic and metabolomic profiling of pPAFAH in in vivo (mouse) and in situ (cultured cell) models of oxidative stress to identify potential metabolic pathway involvement. We will also continue to investigate ML256 and its analogs as tools for structural studies of pPAFAH active site architecture.
5. References
- 1.
- Ames BN. Dietary carcinogens and anticarcinogens. Oxygen radicals and degenerative diseases. Science. 1983;221(4617):1256–64. [PubMed: 6351251]
- 2.
- Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 1990;186:1–85. [PubMed: 2172697]
- 3.
- Harman D. The aging process. Proc. Natl. Acad. Sci. U. S. A. 1981;78(11):7124–8. [PMC free article: PMC349208] [PubMed: 6947277]
- 4.
- Kono N, et al. Protection against oxidative stress-induced hepatic injury by intracellular type II platelet-activating factor acetylhydrolase by metabolism of oxidized phospholipids in vivo. J. Biol. Chem. 2008;283(3):1628–36. [PubMed: 18024956]
- 5.
- Southorn PA, Powis G. Free radicals in medicine. II. Involvement in human disease. Mayo. Clin. Proc. 1988;63(4):390–408. [PubMed: 3280885]
- 6.
- Fruhwirth GO, Loidl A, Hermetter A. Oxidized phospholipids: from molecular properties to disease. Biochim. Et Biophys. Acta. 2007;1772(7):718–36. [PubMed: 17570293]
- 7.
- Kinnunen PK. On the principles of functional ordering in biological membranes. Chem. Phys. Lipids. 1991;57(2–3):375–99. [PubMed: 2054913]
- 8.
- Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog. Lipid Res. 2003;42(4):318–43. [PubMed: 12689622]
- 9.
- Nigam S, Schewe T. Phospholipase A(2)s and lipid peroxidation. Biochim. Et Biophys. Acta. 2000;1488(1–2):167–81. [PubMed: 11080686]
- 10.
- Blank ML, et al. A specific acetylhydrolase for 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine (a hypotensive and platelet-activating lipid). J. Biol. Chem. 1981;256(1):175–8. [PubMed: 7451433]
- 11.
- Farr RS, et al. Preliminary studies of an acid-labile factor (ALF) in human sera that inactivates platelet-activating factor (PAF). Clin. Immunol. Immunopathol. 1980;15(3):318–330. [PubMed: 7371225]
- 12.
- Zimmerman GA, et al. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit. Care Med. 2002;30(5 Suppl):S294–301. [PubMed: 12004251]
- 13.
- Prescott SM, et al. Platelet-activating factor and related lipid mediators. Annu. Rev. Biochem. 2000;69:419–45. [PubMed: 10966465]
- 14.
- Anderson JL. Lipoprotein-associated phospholipase A2: an independent predictor of coronary artery disease events in primary and secondary prevention. Am. J. Cardiol. 2008;101(12A):23F–33F. [PubMed: 18549868]
- 15.
- Sudhir K. Clinical review: Lipoprotein-associated phospholipase A2, a novel inflammatory biomarker and independent risk predictor for cardiovascular disease. J. Clin. Endocrinol. Metab. 2005;90(5):3100–5. [PubMed: 15713711]
- 16.
- Wilensky RL, Macphee CH. Lipoprotein-associated phospholipase A(2) and atherosclerosis. Curr. Opin. Lipidol. 2009;20(5):415–20. [PubMed: 19667981]
- 17.
- Karabina SA, Ninio E. Plasma PAF-acetylhydrolase: an unfulfilled promise? Biochim. Et Biophys. Acta. 2006;1761(11):1351–8. [PubMed: 16807087]
- 18.
- Jessani N, et al. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc. Natl. Acad. Sci. U. S. A. 2002;99(16):10335–40. [PMC free article: PMC124915] [PubMed: 12149457]
- 19.
- Leung D, et al. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat. Biotechnol. 2003;21(6):687–91. [PubMed: 12740587]
- 20.
- Bachovchin DA, et al. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat. Biotechnol. 2009;27(4):387–94. [PMC free article: PMC2709489] [PubMed: 19329999]
- 21.
- Blackie JA, et al. The identification of clinical candidate SB-480848: a potent inhibitor of lipoprotein-associated phospholipase A2. Bioorg. Med. Chem. Lett. 2003;13(6):1067–70. [PubMed: 12643913]
- 22.
- Li X, et al. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab. Dispos. 2009;37(6):1242–50. [PMC free article: PMC3202349] [PubMed: 19282395]
- 23.
- Li X, Kamenecka TM, Cameron MD. Bioactivation of the epidermal growth factor receptor inhibitor gefitinib: implications for pulmonary and hepatic toxicities. Chem. Res. Toxicol. 2009;22(10):1736–42. [PubMed: 19803472]
- 24.
- MacCoss MJ, Wu CC, Yates JR 3rd. Probability-based validation of protein identifications using a modified SEQUEST algorithm. Anal. Chem. 2002;74(21):5593–9. [PubMed: 12433093]
- 25.
- Washburn MP, Wolters D, Yates JR 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19(3):242–7. [PubMed: 11231557]
- 26.
- Wolters DA, Washburn MP, Yates JR 3rd. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001;73(23):5683–90. [PubMed: 11774908]
- 27.
- Hsu KL, Cravatt BF. 2012. Manuscript in preparation.
- 28.
- Bachovchin DA, et al. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc. Natl. Acad. Sci. U. S. A. 2010;107(49):20941–6. [PMC free article: PMC3000285] [PubMed: 21084632]
- 29.
- Chang JW, Nomura DK, Cravatt BF. A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem. Biol. 2011;18(4):476–84. [PMC free article: PMC3119342] [PubMed: 21513884]
- 30.
- Chiang KP, et al. An enzyme that regulates ether lipid signaling pathways in cancer annotated by multidimensional profiling. Chem. Biol. 2006;13(10):1041–50. [PubMed: 17052608]
- 31.
- Kathuria S, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003;9(1):76–81. [PubMed: 12461523]
- 32.
- Li W, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J. Am. Chem. Soc. 2007;129(31):9594–5. [PubMed: 17629278]
- 33.
- Long JZ, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009;5(1):37–44. [PMC free article: PMC2605181] [PubMed: 19029917]
- 34.
- Long JZ, et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc. Natl. Acad. Sci. U. S. A. 2009;106(48):20270–5. [PMC free article: PMC2787168] [PubMed: 19918051]
- 35.
- Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem. Biol. 2005;12(11):1179–87. [PMC free article: PMC1994809] [PubMed: 16298297]
- 36.
- Zhang D, et al. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology. 2007;52(4):1095–105. [PubMed: 17217969]
- 37.
- Williams ET, et al. Genomic analysis of the carboxylesterases: identification and classification of novel forms. Mol. Phylogenet. Evol. 2010;57(1):23–34. [PubMed: 20510380]
- 38.
- Roche M, et al. Augmentation of endogenous cannabinoid tone modulates lipopolysaccharide-induced alterations in circulating cytokine levels in rats. Immunology. 2008;125(2):263–71. [PMC free article: PMC2561133] [PubMed: 18393970]
- 39.
- Nomura DK, et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140(1):49–61. [PMC free article: PMC2885975] [PubMed: 20079333]
- 40.
- Schlosburg JE, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010;13(9):1113–9. [PMC free article: PMC2928870] [PubMed: 20729846]
- 41.
- Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009;16(7):744–53. [PMC free article: PMC2867454] [PubMed: 19635411]
- 42.
- Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010;2(6):949–964. [PMC free article: PMC2904065] [PubMed: 20640225]
- 43.
- Potashman MH, Duggan ME. Covalent modifiers: an orthogonal approach to drug design. J. Med. Chem. 2009;52(5):1231–46. [PubMed: 19203292]
- 44.
- Singh J, et al. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011;10(4):307–17. [PubMed: 21455239]
- 45.
- Kodadek T. Rethinking screening. Nat. Chem. Biol. 2010;6(3):162–165. [PubMed: 20154660]
- 46.
- Vincent PW, et al. Anticancer efficacy of the irreversible EGFr tyrosine kinase inhibitor PD 0169414 against human tumor xenografts. Cancer Chemother. Pharmacol. 2000;45(3):231–8. [PubMed: 10663641]
- 47.
- Robertson JG. Mechanistic basis of enzyme-targeted drugs. Biochemistry. 2005;44(15):5561–71. [PubMed: 15823014]
- 48.
- Adibekian A, et al. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat. Chem. Biol. 2011;7(7):469–78. [PMC free article: PMC3118922] [PubMed: 21572424]
- 49.
- Jessani N, et al. A streamlined platform for high-content functional proteomics of primary human specimens. Nat. Methods. 2005;2(9):691–7. [PubMed: 16118640]
- 50.
- Cociorva D, LTD, Yates JR. Validation of tandem mass spectrometry database search results using DTASelect. Curr. Protoc. Bioinformatics. 2007;Chapter 13(Unit 13):4. [PubMed: 18428785]
- 51.
- Mohler ER 3rd, et al. The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: the results of a multicenter, randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol. 2008;51(17):1632–41. [PubMed: 18436114]
- 52.
- Serruys PW, et al. Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118(11):1172–82. [PubMed: 18765397]
- 53.
- O’Donoghue ML, et al. Study design and rationale for the Stabilization of pLaques usIng Darapladib-Thrombolysis in Myocardial Infarction (SOLID-TIMI 52) trial in patients after an acute coronary syndrome. Am. Heart J. 2011;162(4):613–619 e1. [PubMed: 21982651]
- 54.
- Wilensky RL, et al. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat. Med. 2008;14(10):1059–66. [PMC free article: PMC2885134] [PubMed: 18806801]
- 55.
- Berger JS, et al. Peripheral artery disease, biomarkers, and darapladib. Am. Heart J. 2011;161(5):972–8. [PMC free article: PMC3750980] [PubMed: 21570531]
Supplemental Information
%20at%205%20%003BCM%20compound%20concentration%20(AID%20588474).&p=BOOKS&id=133433_ml256f13.jpg "Click on image to zoom")
Figure S1Gel-based competitive ABPP analysis of cherry-picked HTS hits against over-expressed pPAFAH in a complex proteome (293T Hek cell lysate) at 5 μM compound concentration (AID 588474)
See Table S1 for quantified results and compound SID/CID numbers. Fluorescent images shown in grey scale.
Table S1Activity of cherry-picked HTS hits against pPAFAH as assessed by gel-based competitive ABPP (AID 588474)
| Compound lab name | SID | CID | % Inhibition pPAFAH (5 μM Cpd) |
|---|---|---|---|
| A03 | 49818553 | 24817488 | 9% |
| A04 | 49825105 | 4869553 | 36% |
| A05 | 14723122 | 1075762 | 39% |
| A06 | 17413867 | 5734364 | 91% |
| A07 | 49672084 | 2729092 | 82% |
| A08 | 49717433 | 4095299 | 31% |
| A09 | 49646777 | 16406874 | 44% |
| A10 | 49647446 | 4868796 | 34% |
| A11 | 24803039 | 3713423 | −3% |
| A12 | 49716397 | 1506393 | 21% |
| B03 | 865529 | 666788 | 15% |
| B04 | 49647532 | 728803 | 24% |
| B05 | 49735046 | 4351780 | 30% |
| B06 | 7977938 | 1037259 | 40% |
| B07 | 24837627 | 4318214 | 44% |
| B08 | 49677790 | 1582142 | 93% |
| B09 | 24707853 | 5459455 | 84% |
| B10 | 24820189 | 1471280 | −3% |
| B11 | 24823024 | 16010039 | 6% |
| B12 | 4244777 | 3239329 | 14% |
| C03 | 49825219 | 8016642 | 28% |
| C04 | 56463309 | 186136 | 16% |
| C05 | 17414793 | 5337390 | 23% |
| C06 | 22405959 | 2435938 | 30% |
| C07 | 49827389 | 24819624 | 10% |
| C08 | 49717475 | 24789974 | 8% |
| C09 | 4241429 | 3236404 | 13% |
| C10 | 7965604 | 5142655 | 0% |
| C11 | 85198338 | 3557212 | 19% |
| C12 | 4241939 | 3236852 | 11% |
| D03 | 4265651 | 893857 | 23% |
| D04 | 4244235 | 3238857 | 22% |
| D05 | 14735286 | 2901168 | 35% |
| D06 | 14736542 | 1291961 | 43% |
| D07 | 24789783 | 1092909 | 20% |
| D08 | 4262614 | 675677 | 5% |
| D09 | 14727785 | 983012 | 28% |
| D10 | 14746123 | 2251357 | 22% |
| D11 | 49647657 | 8016217 | 26% |
| D12 | 85269573 | 3642677 | 20% |
| E03 | 14730879 | 2227768 | 26% |
| E04 | 49717482 | 4299457 | 18% |
| E05 | 4246105 | 3240490 | 24% |
| E06 | 49719647 | 16031740 | 13% |
| E07 | 3712811 | 847829 | 17% |
| E08 | 85271190 | 3666144 | 36% |
| E09 | 4244707 | 3239272 | 10% |
| E10 | 49825244 | 24818849 | 30% |
| E11 | 57263442 | 7613142 | 7% |
| E12 | 14741589 | 1835517 | 5% |
| F03 | 845873 | 648231 | 12% |
| F04 | 49733222 | 2176514 | 5% |
| F05 | 57257880 | 25162008 | 64% |
| F06 | 24790869 | 5049848 | 10% |
| F07 | 17385991 | 2104350 | 6% |
| F08 | 49820822 | 4868856 | 31% |
| F09 | 4258391 | 973780 | 18% |
| F10 | 49717429 | 24789969 | 18% |
| F11 | 24782606 | 16187915 | 24% |
| F12 | 17385869 | 11839061 | 0% |
| G03 | 26671454 | 2929094 | 15% |
| G04 | 24818607 | 1483231 | 40% |
| G05 | 22409568 | 3270837 | 24% |
| G06 | 49717013 | 7573833 | 18% |
| G07 | 49645759 | 24747337 | 100% |
| G08 | 56323414 | 9314931 | 44% |
| G09 | 57257322 | 25161540 | −9% |
| G10 | 49736277 | 24793740 | 21% |
| G11 | 57265894 | 16307002 | 75% |
| G12 | 14739000 | 2238485 | 8% |
| H03 | 17504315 | 12004490 | 45% |
| H04 | 7967489 | 651937 | 55% |
| H05 | 846032 | 648404 | 65% |
| H06 | 46500928 | 3241208 | 10% |
| H07 | 24791358 | 3271558 | 40% |
| H08 | 7972767 | 807071 | 55% |
| H09 | 56422452 | 28693 | 15% |
| H10 | 4251449 | 3245135 | 8% |
| H11 | 49822987 | 16406937 | 11% |
| H12 | 56324777 | 24983114 | 7% |
| I03 | 57258588 | 25162597 | 41% |
| I04 | 853294 | 655946 | 84% |
| I05 | 4265310 | 977306 | 44% |
| I06 | 49648667 | 16457475 | 23% |
| I07 | 56315773 | 16231989 | 84% |
| I08 | 49732024 | 6462016 | 22% |
| I09 | 17516379 | 5122563 | 56% |
| I10 | 49728290 | 1092926 | 3% |
| I11 | 49728289 | 1092856 | 21% |
| I12 | 4256580 | 1092921 | 7% |
| J03 | 22409711 | 15945323 | 23% |
| J04 | 3714380 | 2998774 | 38% |
| J05 | 49815912 | 24543718 | 55% |
| J06 | 56321507 | 24981655 | 28% |
| J07 | 17432567 | 1345082 | 45% |
| J08 | 56317169 | 24979689 | 16% |
| J09 | 7966421 | 5308219 | 53% |
| J10 | 56324883 | 24983159 | 42% |
| J11 | 26727296 | 2726311 | 99% |
| K03 | 85269967 | 2327104 | 21% |
| K04 | 17431795 | 2282503 | 48% |
| K05 | 14725991 | 1160042 | 43% |
| K06 | 85270454 | 4783698 | 17% |
| K07 | 49819107 | 2213570 | 12% |
| K08 | 49728592 | 1092738 | 69% |
| K09 | 14723216 | 1092878 | 38% |
| K10 | 49672846 | 4876778 | 35% |
| K11 | 17506881 | 2971862 | 8% |
| L03 | 26664791 | 288774 | 29% |
| L04 | 3716333 | 2084085 | 54% |
| L05 | 26669611 | 2972134 | 47% |
| L06 | 56324509 | 24982966 | 24% |
| L07 | 14739800 | 1333712 | 27% |
| L08 | 17508962 | 1150498 | 39% |
| L09 | 4241284 | 1480154 | 8% |
| L10 | 49829001 | 16406654 | 40% |
| L11 | 49732864 | 20968792 | 5% |
| M03 | 14733155 | 4048822 | 52% |
| M04 | 24831877 | 4374113 | 48% |
| M05 | 49823145 | 4869036 | 53% |
| M06 | 49829271 | 4869139 | 19% |
| M07 | 24823472 | 16195494 | 3% |
| M08 | 49726570 | 3152114 | 17% |
| M09 | 26729061 | 2795735 | 12% |
| M10 | 17414795 | 5337401 | 9% |
| M11 | 49665098 | 4869534 | 18% |
| N03 | 14728016 | 847926 | 41% |
| N04 | 14742766 | 1951620 | 70% |
| N05 | 49828442 | 16406741 | 53% |
| N06 | 56315412 | 18108581 | 18% |
| N07 | 14733456 | 3505699 | 18% |
| N08 | 57260648 | 16306230 | 78% |
| N09 | 14731640 | 1298699 | 57% |
| N10 | 49717138 | 16034280 | 4% |
| N11 | 56322366 | 24982068 | 20% |
| O03 | 24819918 | 1486626 | 36% |
| O04 | 7977385 | 749766 | 22% |
| O05 | 49828888 | 17584900 | 73% |
| O06 | 49829242 | 4967664 | 37% |
| O07 | 49820844 | 24818165 | 10% |
| O08 | 49645631 | 24747236 | 29% |
| O09 | 49733753 | 1091834 | 24% |
| O10 | 14739190 | 955142 | 9% |
| O11 | 49645884 | 16406847 | 21% |
| P03 | 4264232 | 2236013 | 79% |
| P04 | 49717421 | 4103720 | 41% |
| P05 | 57263037 | 4592650 | 53% |
| P06 | 17413874 | 2229425 | 46% |
| P07 | 87334149 | 16060009 | 18% |
| P08 | 14723147 | 1008028 | 30% |
| P09 | 4257433 | 1378785 | 98% |
| P10 | 14725052 | 1015202 | 46% |
| P11 | 14737509 | 4048820 | 29% |
Table S2Target SAR Analysis: non-carbamate HTS Hits
| Structure | Potency and Selectivity† | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | AID 588773 | ||||||||||||
| % INH pPAFAH | Anti-targets‡ | Fold Selectivity | |||||||||||
| Entry | Lab Name | SID | CID | SRID | (mouse brain membrane) | ||||||||
| 10 μM | 1 μM | 0.1 μM | 10 μM | 1 μM | 0.1 μM | ||||||||
| G07* | 49645759 | 24747337 | SR-01000781101-2 |
 | ND | 93 | 88 | 33 | FAAH, MAGL, ABHD6 | FAAH | none | 0 | |
| 10 | G11* | 57265894 | 16307002 | SR-01000863254-2 |
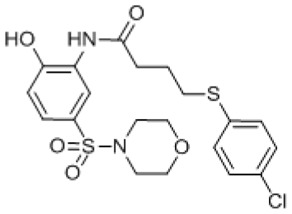 | ND | 88 | 50 | 18 | none | none | none | >10 |
| 11 | N08* | 57260648 | 16306230 | SR-01000863326-2 |
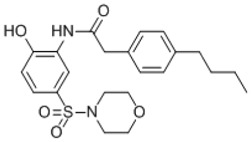 | ND | 67 | 15 | 0 | none | none | none | >1 |
| 12 | I07* | 56315773 | 16231989 | SR-01000843990-2 |
 | ND | 34 | 0 | 0 | none | none | none | ND |
- *
MLSMR library compounds
- †
Color scheme: green = ≥ 50% inhibition pPAFAH, or ≥10-fold selective; grey = not determined (ND); orange = one or more anti-target(s) with >50% INH
- ‡
Anti-targets: FAAH: fatty acid amide hydrolase; ABHD6: abhydrolase domain containing protein 6; MAGL: monoacylglycerol lipase
Table S3Serine Hydrolase (SH) Abbreviations and Names
| Abbreviation | Name | GeneID | Protein GI |
|---|---|---|---|
| pPAFAH (Pla2g7) | plasma platelet activating factor acetylhydrolase | 27226 | 31980752 |
| Abhd3 | abhydrolase domain containing protein 3 | 106861 | 19527360 |
| Abhd4 | abhydrolase domain containing protein 4 | 105501 | 326937491 |
| Abhd6 | abhydrolase domain containing protein 6 | 66082 | 31560264 |
| Abhd10 | abhydrolase domain containing protein 10 | 213012 | 269784760 |
| Abhd11 | abhydrolase domain containing protein 11 | 68758 | 21644577 |
| Abhd12 | abhydrolase domain containing protein 12 | 76192 | 159110817 |
| Ache | acetylcholinesterase | 11423 | 13928664 |
| Acot1 | acyl-coenzyme A thioesterase 1 | 26897 | 6753550 |
| Acot2 | acyl-coenzyme A thioesterase 2 | 171210 | 238624114 |
| Acot3 | acyl-coenzyme A thioesterase 3 | 171281 | 19527406 |
| Acot6 | acyl-coenzyme A thioesterase 6 | 217700 | 110626167 |
| Acot7 | acyl-coenzyme A thioesterase 7 | 70025 | 225690616 |
| Apeh | acylpeptide hydrolase | 235606 | 19343726 |
| Bat5 | abhydrolase domain-containing protein 16A | 193742 | 30519896 |
| Dpp8 | dipeptidyl peptidase 8 | 74388 | 31542571 |
| Dpp9 | dipeptidyl peptidase 9 | 224897 | 255003757 |
| Ces1 (Es1) | liver carboxylesterase 1 | 13884 | 247269929 |
| Esd | esterase D/formylglutathione hydrolase | 13885 | 13937355 |
| Faah | fatty acid amide hydrolase | 14073 | 123253900 |
| Fam108a | family with sequence similarity 108, member A | 216169 | 21703840 |
| Fam108b1 | family with sequence similarity 108, member B1 | 226016 | 38142456 |
| Fam108c1 | family with sequence similarity 108, member C1 | 70178 | 158186616 |
| Fasn | fatty acid synthase | 14104 | 93102409 |
| Kiaa1363 (Nceh1; Aadacl1) | neutral cholesterol ester hydrolase 1 | 320024 | 30520239 |
| Lactb | lactamase, beta | 80907 | 13507666 |
| Lipe | hormone-sensitive lipase | 16890 | 87239972 |
| Lypla1 | lysophospholipase 1 | 18777 | 6678760 |
| Lypla2 | lysophospholipase 2 | 26394 | 7242156 |
| Lyplal1 | lysophospholipase-like 1 | 226791 | 227496223 |
| Magl (Mgll) | monoacylclycerol lipase | 23945 | 261878511 |
| Nceh1 (Aadacl1; Kiaa1363) | neutral cholesterol ester hydrolase 1 | 320024 | 30520239 |
| Pafah1b2 | platelet-activating factor acetylhydrolase IB subunit beta | 18475 | 40254624 |
| Pafah1b3 | platelet-activating factor acetylhydrolase IB subunit gamma | 18476 | 6679201 |
| Parl | presenilins-associated rhomboid-like protein | 381038 | 54261813 |
| Pla2g15 | group XV phospholipase A2 precursor | 192654 | 19527008 |
| Pnpla6 | patatin-like phospholipase domain containing protein 6 | 50767 | 170763472 |
| Pnpla8 | patatin-like phospholipase domain containing protein 8 | 67452 | 118130807 |
| Pme1 (Ppme1) | protein phosphatase methylesterase 1 | 72590 | 30794138 |
| Prcp | proline carboxypeptidase | 72461 | 33469015 |
| Prep | prolyl endopeptidase | 19072 | 6755152 |
| Prepl | prolyl endopeptidase-like | 213760 | 254939518 |
| Rbbp9 | retinoblastoma binding protein 9 | 26450 | 86439977 |
| Tpp2 | tripeptidyl peptidase 2 | 22019 | 37194903 |
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Review Optimization and characterization of a triazole urea inhibitor for platelet-activating factor acetylhydrolase type 2 (PAFAH2).[Probe Reports from the NIH Mol...]Review Optimization and characterization of a triazole urea inhibitor for platelet-activating factor acetylhydrolase type 2 (PAFAH2).Adibekian A, Hsu KL, Speers AE, Monillas ES, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Bahnson BJ, Cravatt BF, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Optimization and characterization of a triazole urea inhibitor for diacylglycerol lipase beta (DAGL-β).[Probe Reports from the NIH Mol...]Review Optimization and characterization of a triazole urea inhibitor for diacylglycerol lipase beta (DAGL-β).Hsu KL, Tsuboi K, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Cravatt BF, Hodder P, Rosen H. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Optimization and characterization of a triazole urea inhibitor for alpha/beta hydrolase domain-containing protein 11 (ABHD11): anti-probe for LYPLA1/LYPLA2 dual inhibitor ML211.[Probe Reports from the NIH Mol...]Review Optimization and characterization of a triazole urea inhibitor for alpha/beta hydrolase domain-containing protein 11 (ABHD11): anti-probe for LYPLA1/LYPLA2 dual inhibitor ML211.Adibekian A, Hsu KL, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Cravatt BF, Hodder P, Rosen H. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Probe Development Efforts to Identify Novel Inhibitors of ABHD10.[Probe Reports from the NIH Mol...]Review Probe Development Efforts to Identify Novel Inhibitors of ABHD10.Zuhl AM, Mohr JT, Speers AE, Bachovchin DA, Berlin JM, Spicer T, Fernandez-Vega V, Brown SJ, Ferguson J, Fu GC, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Optimization and characterization of a triazole urea dual inhibitor for lysophospholipase 1 (LYPLA1) and lysophospholipase 2 (LYPLA2).[Probe Reports from the NIH Mol...]Review Optimization and characterization of a triazole urea dual inhibitor for lysophospholipase 1 (LYPLA1) and lysophospholipase 2 (LYPLA2).Adibekian A, Martin BR, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Cravatt BF, Hodder P, Rosen H. Probe Reports from the NIH Molecular Libraries Program. 2010
- Optimization and characterization of a carbamate inhibitor for plasma platelet-a...Optimization and characterization of a carbamate inhibitor for plasma platelet-activating factor acetylhydrolase (pPAFAH) - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...