

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

A critical and dynamic epigenetic post-translational modification involves Nε-methylation of histone lysine residues by histone methyltransferases. This process was originally thought to be an irreversible epigenetic mark, yet two representative classes of histone lysine demethylases which reverse this process are LSD1/2 and the Jumonji domain containing proteins (JMJD) have emerged. Despite an increased interest in these enzymes as a result of their suspected role in a variety of diseases (e.g. cancer and virus infection), a dearth of potent and cell-permeable inhibitors of the JMJD2 enzymes remain. As such, we sought to discover novel small molecule inhibitors of the JMJD2 family of histone demethylases via a quantitative high throughput screen and subsequent medicinal chemistry optimization campaign. Herein, we describe the discovery and optimization of N-(3-(dimethyamino)propyl-4-(8-hydroxyquinolin-6-yl)benzamide, ML324, a probe molecule that displays submicromolar inhibitory activity toward JMJD2E (in vitro) and possesses excellent in vitro ADME properties. In contrast to previously reported inhibitors of the JMJD proteins, ML324 displays excellent cell permeability providing an opportunity for more extensive cell-based studies of JMJD2 enzymes to be undertaken. In addition, ML324 demonstrates potent anti-viral activity against both herpes simplex virus (HSV) and human cytomegalovirus (hCMV) infection via inhibition viral IE gene expression. ML324 suppresses the formation of HSV plaques, even at high MOI, and blocks HSV-1 reactivation in a mouse ganglia explant model of latently infected mice. The studies described herein provide the basis for the use of JMJD2 inhibitors in proof-of-concept animal models for treatment of herpes virus infections and recurrence.
Assigned Assay Grant #: MH084681
Screening Center Name & PI: NIH Chemical Genomics Center, John McKew
Chemistry Center Name & PI: NIH Chemical Genomics Center, John McKew
Assay Submitter & Institution: Udo Oppermann, Structural Genomics Consortium, University of Oxford
PubChem Summary Bioassay Identifier (AID): 2421
Probe Structure & Characteristics

JMJD Inhibitor
| CID/ML# | Target Name | IC50/EC50 (nM) [SID, AID] | Anti-target Name(s) | IC50/EC50 (μM) [SID, AID] | Fold Selective | Secondary Assay(s) Name: IC50/EC50 (μM) [SID, AID] |
|---|---|---|---|---|---|---|
| ML324/CID 44143209 | JMJD2E | 920 nM (JMJD2E) | LSD1 | Inactive [SID 85736407, AID 687009] | > 100-fold | LSD1 [SID 85736407, AID 687008] |
Recommendations for scientific use of the probe
ML324, with its potent inhibition and favorable ADME properties (e.g. solubility, cell permeability, microsomal stability) allows researchers to study the modulation of the JMJD2 family of histone demethylases in vivo. Previous reports have demonstrated the importance of the JMJD2 family of histone demethylases in a variety of important biological processes and diseases (e.g. cancer). In this report, ML324 was used in proof-of-concept studies investigating the role of the JMJD2 proteins in initiation of HSV and hCMV infections and was found to display potent anti-viral activity. Given this promising activity, ML324 could be used in more extensive investigations of the mechanisms involved in chromatin regulation of HSV and other related viruses both in a cellular context and in vivo. Moreover, ML324 could be used to study the role of JMJD2 proteins in the context of cancer biology.
1. Introduction
Epigenetic modifications, such as inherited and acquired modifications of DNA and histones, affect the regulation of gene expression, chromosome stability, genomic imprinting, and stem cell fate. More specifically, epigenetic modifications of DNA methylation and histone modifications can consist of acetylation, methylation, phosphorylation, ubiquitination, glycosylation, and sumoylation [1] and these changes can occur at single or multiple sites and in various combinations. The interplay among methylation states and other histone modifications to direct transcriptional outcome is commonly referred to as the histone code. Given the wide-spread importance of chromatin regulation in cell biology, and progression of human disease, the proteins involved in these critical post-translational modifications have attracted much interest from the academic and pharmaceutical sectors alike. Accordingly, there has been significant progress in the realm of DNA methylation and histone deacetylase inhibitors (HDACi), with the FDA approval of 5-azacytidine and decitabine (DNA methylating agents) and vorinostat and valproic acid (HDACi). However, little clinical progress has been made toward the development of histone demethylase or methyl transferase inhibitors, largely because the cellular functions of these enzymes remain poorly understood. One challenge is that, unlike histone acetylation/deacetylation which alters the charge of the histone tail, histone methylation/demethylases influences the basicity, hydrophobicity, and affinity of molecules such as transcription factors toward DNA. Accordingly, histone acetylation typically results in transcription activation and histone deacetylation leads to gene silencing and the effect of histone demethylases and methyltransferease is highly dependent on the site of methylation and the context in the genome.
Histone methylation regulates fundamental biological processes, such as X chromosome inactivation, heterochromatin formation, transcriptional regulation, genomic imprinting, and DNA repair. [2] Importantly, almost all cancers show altered chromatin modifications and dysregulation of chromatin modulating enzymes, with histone lysine acetylation and methylation being the most prominent posttranslational modifications. In addition, many nuclear virus-pathogens such as herpes simplex virus (HSV) and human cytomegalovirus (hCMV) must contend with and utilize host cell chromatin for successful infection, regulation of viral latency, and recurrent reactivation [3] In fact; previous studies have demonstrated the use of epigenetic inhibitors, and more specifically inhibitors of histone demethylase LSD1, for targeting the initiation of viral infection. [4] However, LSD1 only removes H3K9 mono and di-methylation. Demethylation of the tri-methylated H3K9 is accomplished by the Jumonji-domain-containing proteins (JMJD) enzymes which represent the largest class of N ε -methyl lysine demethylases, and utilize non-heme Fe(II) in an α-ketoglutarate-dependent dioxygenation mechanism. Thus, these enzymes actively participate in regulation of gene transcription via antagonizing the action of a class of repressive histone methyltransferases. Of the JMJD family of enzymes, JMJD2E in particular emerged as the first candidate for high-throughput screening (HTS) and probe development due to its stability and high specific activity. [5] However, to date, only compounds which possess weak inhibition and poor cell permeability have been reported; examples include 2,4-pyridine dicarboxylic acid (PDCA) [6], N-oxalylglycine (oxoglutarate mimetic) [7], and analogs thereof (see prior art section). The lack of high-quality inhibitors of the JMJD2 family of enzymes necessitated a search for optimizable chemotypes which possess sufficient cell permeability to allow for cell-based studies involving this important protein target.
Prior Art Analysis: Inhibitors of JMJD demethylases
At the onset of this probe development program, few small molecule JMJD2 inhibitors were reported in the literature (Figure 1). 2,4-PDCA [6] and dimethyl ester of N-oxalylglycine (DMOG) [7] are among the first reported JMJD2 inhibitors. Both these compounds are non-selective JMJD2 inhibitors with limited cell activity. During the course of our program, other small molecule inhibitors, mainly analogs derived from 2,4-PDCA and N-oxalyl glycine core were reported. Among them, Thalhammer and co-workers reported 3-amino-2,4-PDCA [8], a potent JMJD2E inhibitor derived from 2,4-PDCA. Small peptide like molecules P-1 [9], P-2 [10] and the N-oxalyl-d-tyrosine analog [11] were reported as selective JMJD2 inhibitors. Recently, GSK reported a very potent inhibitor, GSK-J1 [12], selective for JMJD3 and thus has no activity against JMJD2 enzymes. Moreover, this compound has very poor permeability and even the prodrug ester has only modest activity in cell-based studies. Our own efforts resulted in the discovery of the JMJD2E inhibitor 8-HQ-5-COOH (1) [5] which is the basis for our SAR exploration described herein. All of these existing compounds possess a carboxylic acid functionality which is necessary for the potency but contributes towards poor cell permeability. Moreover, existing compounds lack more drug-like characteristics with desired molecular and physicochemical properties to investigate in cell based assays and animal models. Thus, ML324 is the first potent and cell permeable JMJD2 inhibitor with desirable in vitro ADME properties, and represents an excellent starting point for further optimization.
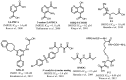
Figure 1
Previously reported inhibitors of JMJD histone demethylases.
2. Materials and Methods
General Methods for Chemistry. All air or moisture sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Anhydrous solvents such as dichloromethane, N,N-dimethylforamide (DMF), acetonitrile, methanol and triethylamine were purchased from Sigma-Aldrich. Preparative purification was performed on a Waters semi-preparative HPLC system. The column used was a Phenomenex Luna C18 (5 micron, 30 × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10% to 50% acetonitrile over 8 minutes was used during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA). Method 1: A 7 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 minute run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50 °C. Method 2: A 3 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 minute run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 micron, 3 × 100 mm) was used at a temperature of 50 °C. Purity determination was performed using an Agilent Diode Array Detector for both Method 1 and Method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical shifts are reported in ppm with undeuterated solvent (DMSO-d6 at 2.49 ppm) as internal standard for DMSO-d6 solutions. All of the analogs tested in the biological assays have purity greater than 95%, based on both analytical methods. High resolution mass spectrometry was recorded on Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
2.1. Assays
2.1.1. JMJD2E qHTS FDH Assay
Enzyme and buffer solutions (3 μL) were dispensed into a 1,536-well Greiner black solid-bottom assay plate. The library compounds (23 nL) were transferred using a Kalypsys pintool equipped with 1,536-pin array. The plate was incubated at room temperature (15 min), and then a 1 μL aliquot of substrate solution was added to initiate the reaction. The plate was transferred to ViewLux imager where an initial reading using standard UV optics (Ex 340 nm, Em 450 nm) was obtained. The plate was then removed from the reader, incubated for 30 minutes at room temperature, and returned to the reader for a second fluorescence reading. A fully automated robotic screening system (Kalypsys Inc, San Diego, CA) was used to perform the above steps as described previously. Compound plates containing DMSO as a vehicle-only control were included at regular interval throughout the screen to monitor any systematic trend in the assay signal associated with reagent dispenser variation or decreases in enzyme specific activity. For activity calculations, percent values were computed as the difference in fluorescence intensity between last and first time points. The percentage activity was calculated from the median values of the catalyzed, or neutral control, and the uncatalyzed, or 100% inhibited, control, respectively, using in-house software (http://ncgc.nih.gov/pub/openhts/).
2.1.2. MALDI-TOF-MS Assay (AID: 2688)
Matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF-MS) assay for JMJD2E activity (hit validation). For the MS assays, JMJD2E (2 μM), FAS (10 μM) and SA (100 μM) in HEPES buffer, 50 mM, pH 7.5, were incubated with inhibitor (stock solutions in DMSO, final in-assay concentration varied, but final DMSO concentration was 5 % of assay mix) for 15 min at room temperature. Disodium 2OG (10 μM) and peptide (10 μM) were added, and the mixture was incubated for 30 min at 37 °C, before quenched with 1:1 methanol, followed by addition of four volumes of 20 mM triammonium citrate. The diluted assay mixture (1 μL) was then mixed with alpha-cyano-4-hydroxycinnamic acid (the MALDI-TOF-MS matrix, 1μL) and spotted onto a MALDI-TOF-MS plate before analysis. The relative intensities of different methylation states observed in the mass spectra were then used to calculate percentage demethylation, and IC50s were calculated from the variation in percentage demethylation at different inhibitor concentrations. Because the sensitivity of this assay was lower than that of the coupled HTS assay (due to its use of higher concentration of enzyme and substrate), this confirmatory assay was only used to qualify hit series for further optimization (Yes/No fashion) but not to drive SAR. Of the 44 synthesized compounds tested in the MALDI-TOF-MS assay, 41 were active and 3 were inconclusive (>100 uM IC50). Compound 1 gave an IC50 of 2.4 μM (see Figure 8).
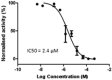
Figure 8
Concentration response curve of compound 1 (CID: 459617) in MALDI-TOF-MS hit validation assay.
2.1.3. AlphaScreen Assay
Inhibition of JMJD2E was assessed using the histone demethylase AlphaScreen (PerkinElmer) assay. This assay uses a biotinylated peptide substrate and relies on detection of the product methyl mark using a specific antibody coupled to protein-A acceptor beads and a Steptavidin donor bead to capture the peptide. The assay was performed in 384-well plate format using white proxiplates (Perkin Elmer). All steps were carried out in assay buffer (50 mM HEPES pH 7.5, 0.1% (w/v) BSA and 0.01 % (v/v) Tween-20). FAS was dissolved fresh each day in 20 mM HCl to a concentration of 400 mM and diluted to 1 mM in deionized water. All other components were dissolved fresh each day in deionized water. For IC50 determinations, 4.9 μL of assay buffer containing JMJD2E enzyme (2 nM final concentration) was transferred to wells of a 384-well proxiplate. Titrations of compound (0.1 μL) were transferred to each well and the enzyme was allowed to pre-incubate for 15 min with compound (final concentration of DMSO was 1%). The enzyme reaction was initiated by addition of 5 μL of a substrate mix consisting of 2-OG (10 μM final concentration), FAS (1 μM final concentration), L-Ascorbic Acid (100 μM final concentration) and biotinylated peptide substrate (30 nM final concentration) and the reaction allowed to proceed for 20 min at room temperature. The enzyme reaction was stopped by addition of 5 μL of EDTA (7.5 mM final concentration in assay buffer). Streptavidin Donor beads (0.08 mg/mL) and Protein-A conjugated acceptor beads (0.08 mg/mL) were pre-incubated for 1 hr with anti-H3-K9Me2 antibody (0.2 μg/mL, Abcam). The presence of biotin-H3-product was detected by addition of 5 μL of the pre-incubated AlphaScreen beads (final concentrations of 0.02 mg/mL with respect to acceptor and donor beads). Detection was allowed to proceed for 1 hr at room temperature and the assay plates were read on a BMG Pherastar FS plate reader. Data were normalized to the no enzyme control and the IC50 was determined from the nonlinear regression curve fit using GraphPad Prism 5.
2.1.5. Immunofluorescence
The expression of UL29 in HSV-1 infected MRC-5 and Vero cells was detected using standard immunofluorescence methods.
2.1.6. Ganglia Immunofluorescence
HSV-1 latently infected mouse trigeminal ganglia were explanted and cultured with DMEM-10% FBS in the presence or absence of the indicated compound for 48 hr. Ganglia were then fixed with 4% paraformaldehyde for 12 hr, washed in PBS/70% EtOH, and embedded in paraffin. Sections were de-paraffined with Xylene and rehydrated in a series of Ethanol-H2O solutions. For antigen retrieval, sections were boiled in 0.01 M Citric Acid (pH 6.0) for 10 min, cooled to 48 °C, and equilibrated in PBS. Sections were costained with anti-UL29 sera (gift of W. Ruyechan, SUNY Buffalo) and anti-neurofilament 200 (Sigma, # N0142, 1:200) followed by Donkey anti-mouse IgG-FITC (Jackson ImmunoResearch, #715-095-150) and Donkey anti-Rabbit IgG-TRITC (Jackson ImmunoResearch, #711-025-152). Sections were visualized using a Leica TCS SF5 confocal microscope.
2.1.6. Latently infected trigeminal ganglia
Balb/c mice were infected with 5 × 105 PFU HSV-1 (strain F) per eye. Trigeminal ganglia of latently infected mice were harvested 30 days after clearance of the primary infection. Viral yields from paired ganglia analyses and from drug reversals were performed as described previously. [4] For mRNA levels, cDNA was prepared from paired latently infected ganglia explanted in the presence of Acyclovir (ACV), ML324 or DMOG for 6 hr and quantitated using the primer sets listed in the appendix. Animal care and handling was done in accordance with the NIH Animal Care and Use Guidelines and the NIAID Animal Care and Use Committee.
2.1.7. Inhibition of viral infection in cell culture
Cells were treated with DMSO, LSD1 inhibitor (TCP, tranylcypromine, Sigma P8511), or JMJD2 inhibitors (DMOG, dimethyloxallyl glycine, Cayman 71210; ML324) and infected with HSV-1 or hCMV as described below. cDNA was produced from total RNA and quantitated using an ABI 7900HT (ABI SDS 2.3 Software). Viral yields were determined by titration.
2.1.8. LSD1 Histone Demethylase Assay (AID 687009)
The assay was performed in a buffer containing 10 mM HEPES, Na pH 7.5, 5 mM MgCl2, 50 mM KCl, 0.01% Nonidet P-40, and 0.1% BSA. LSD1 was sourced from BPS Bioscience (Cat. No. 50100, San Diego, CA) and the substrate peptide was synthesized and purified by Tufts University Core Facilities (Boston, MA). Reagents (3 μL) containing either buffer-only (inhibited control) or LSD1 (120 nM) were dispensed into a 1,536-well Aurora black solid bottom cycloolefin plate (see Table 1 for protocol steps). Compounds (23 nL) were transferred via Kalypsys pintool equipped with a 1,536-pin array. The plate was incubated for 10 min at room temperature, followed by the addition of 1 μL 4 X substrate (10 mM HEPES, pH 7.5, 8 U/mL HRP, 800 μM Amplex Red, and 200 μM H3K4Me2 peptide [Sequence H-ARTXQTARKSTGGKAPRKQLA-NH2, where X = Nε,Nε-dimethyl-L-lysine]) to start the reaction. The plate was then centrifuged at 1,000 rpm for 15 seconds, and the fluorescence intensity was recorded on a ViewLux High-throughput CCD imager (Perkin-Elmer) using standard TAMRA optics (525 nm excitation and 598 nm emission). The plate was then incubated for 30 min at room temperature, and a second read on the ViewLux was performed. The fluorescence intensity difference over the 30 min was used to calculate the respective reaction rate for each well.
Table 1
LSD1 histone demethylaseassay protocol.
2.2. Probe Chemical Characterization

Probe Characterization of ML324
*Purity >98% as determined by LC/MS and 1H NMR analyses
ML324: N-(3-(dimethylamino)propyl)-4-(8-hydroxyquinolin-6-yl)benzamide
(NCGC00183808): LC-MS Retention Time: t1 (Method 1) = 3.033 min and t2 (Method 2) = 2.42 min; 1H NMR (400 MHz, DMSO-d6) δ 1.69 (m, 2H), 2.19 (s, 6H), 2.33 (t, J = 7.1 Hz, 2H), 3.27 – 3.38 (m, 2H), 7.46 (d, J = 1.9 Hz, 1H), 7.59 (dd, J = 8.3, 4.2 Hz, 1H), 7.79 (d, J = 1.9 Hz, 1H), 7.93 – 7.85 (m, 2H), 8.02 – 7.94 (m, 2H), 8.40 (dd, J = 8.4, 1.7 Hz, 1H), 8.59 (t, J = 5.6 Hz, 1H), 8.86 (dd, J = 4.2, 1.6 Hz, 1H), 10.04 (s, 1H); 13C NMR (400 MHz, DMSO-d6) δ 165.65, 153.84, 148.47, 142.10, 138.21, 138.11, 136.50, 133.70, 128.98, 127.80, 126.78, 122.37, 115.79, 110.23, 56.83, 45.02, 37.70, 26.94; HRMS (ESI) m/z (M+H)+ calcd. for C21H24N3O2, 350.1863; found 350.1867.

Figure 21H NMR spectrum of ML324

Figure 313C NMR spectrum of ML324
MLS numbers for probe analogs
| Internal ID | MLS ID | SID | CID | ML # | Type | Source |
|---|---|---|---|---|---|---|
| NCGC00183808 | MLS004685729 | 85736407 | 44143209 | ML324 | Probe | NCGC |
| NCGC00183806 | MLS004685730 | 85736405 | 44143205 | Analog | NCGC | |
| NCGC00241035 | MLS004685731 | 104224538 | 49852977 | Analog | NCGC | |
| NCGC00183807 | MLS004685732 | 85736406 | 44143207 | Analog | NCGC | |
| NCGC00244531 | MLS004685733 | 68740002 | 156647495 | Analog | NCGC | |
| NCGC00189502 | MLS004685734 | 104224401 | 49853311 | Analog | NCGC |

Figure 4Structures of the five analogs that have been submitted to the MLSMR
%20DPBS%20buffer%20(pH%207.4)%20b)%20JMJD2E%20AlphaScreen%20assay%20buffer%20(1%20M%20HEPES%20buffer)%20c)%20pH%202%20buffer%20and%20d)%20pH%2010%20buffer.&p=BOOKS&id=169450_ml324f8.jpg "Click on image to zoom")
Figure 5Stability of ML324 measured as percent composition of probe molecule in aqueous solution at r.t. over 48 hr in a) DPBS buffer (pH 7.4) b) JMJD2E AlphaScreen assay buffer (1 M HEPES buffer) c) pH 2 buffer and d) pH 10 buffer

Scheme 1Synthetic route for analogs containing 5-COOH

Scheme 2Synthetic route to ML324
2.3. Probe Preparation
Preparation of ML324
Steps A and B
A mixture of concentrated sulfuric acid (18.5 mL, 347 mmol, 7 eq), water (14.4 mL, 799 mmol), sodium 3-nitrobenzenesulfonate (16.5 g, 77 mmol, 1.5 eq), and glycerol (5.6 mL, 77 mmol, 1.5 eq) was warmed gently to 65 °C with stirring. To this warm mixture, 4-bromo-2-methoxyaniline (10 g, 49.5 mmol, 1 eq) was added in portions. The reaction mixture was stirred at 150 °C for 12 hr. Upon cooling, the reaction mixture was poured into water and neutralized with ammonium hydroxide. The product was extracted with dichloromethane. The organic layer was washed with saturated bicarbonate solution, brine and dried with MgSO4, and concentrated in vacuo. The crude product was purified on a biotage flash® system eluting with 5 % methanol in dichloromethane.
The above product was suspended in concentrated hydrobromic acid (200 mL) and refluxed for 4 days. The reaction mixture was concentrated and neutralized with ammonium hydroxide. The precipitate was collected by filtration and dried under vacuum to get light yellow solid (Yield: 8.3 g; 75 % over 2 steps).
Step C
A 40 % solution of diethyl azodicarboxylate in toluene (22 mL, 55.6 mmol, 1.5 eq) was slowly added to a mixture of 6-bromoquinolin-8-ol (8.3 g, 37.0 mmol, 1 eq), 2-(trimethylsilyl)ethanol (7.93 mL, 55.6 mmol, 1.5 eq), and triphenylphosphine (14.6 g, 55.6 mmol, 1.5 eq) in THF (37.0 mL) and toluene (37.0 mL) at 0 °C. The reaction mixture was slowly warmed to room temperature and stirred for 2 hr. The crude product obtained after concentration was purified on a biotage flash® system eluting with 20 % ethyl acetate in hexanes (yield: 86 %; 10.3 g).
Step D
A mixture of 6-bromo-8-(2-(trimethylsilyl)ethoxy)quinoline (0.25 g, 0.77 mmol, 1 eq), N-(3-(dimethylamino)propyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzamide (0.384 g, 1.16 mmol, 1.5 eq) and a 2 M solution of sodium carbonate (0.96 mL, 1.93 mmol, 2.5 eq) and Pd(PPh3)4 (0.045 g, 0.039 mmol, 5 mol %) in dimethoxyethane was degassed with argon for 5 minutes. The reaction mixture was heated at 150 ºC for 30 min in a Biotage Initiator® microwave reactor. Upon completion of the reaction, the solvent was removed under stream of air and the crude mixture was suspended in methanol/methylene chloride and filtered through a Pd scavenger cartridge. The crude product was purified on a biotage flash® system eluting with 10 % methanol in dichloromethane to get pure product (yield: 84 %; 0.29 g). The pure product was dissolved in dichloromethane (3 mL) and trifluoroacetic acid (2 mL) and heated at 100 ºC for 10 min in a Biotage Initiator® microwave reactor. The solvent was removed under stream of air and the crude product was purified in HPLC (gradient 20-100% acetonitrile w/ 0.1% TFA in water w/ 0.1% TFA) to yield, after lyophilization, pure N-(3-(dimethylamino)propyl)-4-(8-hydroxyquinolin-6-yl)benzamide (ML324) as a TFA salt.
3. Results
3.1. Summary of Screening Results
In preparation for the full-collection screen, we performed a pilot screen of the LOPAC1280 collection using standalone equipment; this was followed by a triplicate LOPAC1280 screen using the fully-integrated Kalypsys robotic system. In both cases, excellent Z' was noted; in addition, the triplicate concentration-response curves from the robotic validation overlapped almost completely. A qHTS of 226,938 samples across 1,316 plates resulted in a high average Z' of 0.85 (Figure 6). Each sample's readout was captured in a kinetic mode with multiple reads per well, including the first background fluorescent read. This robust and stable screen resulted in a large high quality concentration-response dataset that yielded inhibitors with potencies ranging from <100 nM to >57 μM.

Figure 6
Plate Z' trend for the JMJD2E primary qHTS.
Identification of lead
A diverse collection of ∼236,000 compounds was screened in concentration-response format by using a real-time fluorogenic coupled assay designed to monitor formaldehyde production from the demethylation reaction: this assay detects formaldehyde released from the demethylase reaction by converting it to formic acid using formaldehyde dehydrogenase; in turn, the oxidation of formaldehyde is coupled to the reduction of NAD+ to NADH, which is monitored by fluorescence detection. The assay was miniaturized to 1536-well format and was previously demonstrated to perform robustly under large-scale screening settings. [13] Given that the assay detection was in the UV region of the light spectrum, where small molecules are most likely to fluoresce, the first kinetic read for each sample was used to flag compounds that show high background fluorescence. The delta kinetic read calculations for such inactive samples could lead to a concentration-dependent response due to compound fluorescence rather than putative inhibition. After removal of autofluorescent artifacts, initial screening hits were further prioritized by a combination of cheminformatics to remove compounds with reactive functionalities, counterscreening using the coupling enzyme from the HTS, and orthogonal confirmatory detection of inhibition by mass spectrometric assays [5]. Additional hit validation studies were carried out on a member of the top series identified, 8-hydroxyquinolines, which were shown by crystallographic analyses to inhibit by binding to the active site Fe(II) and to modulate demethylation at the H3K9 locus in a cell-based assay utilizing a transiently transfected JMJD2A and immunofluorescence detection.

Figure 7Work flow for the identification and optimization of small molecule inhibitors of JMJD2
%20in%20the%20AlphaScreen%20secondary%20assay.&p=BOOKS&id=169450_ml324f12.jpg "Click on image to zoom")
Figure 9Concentration response curve of compound 1 (CID: 459617) in the AlphaScreen secondary assay
3.2. Dose response curves for probe

Figure 10Concentration response curve of ML324 (CID-44143209) in the AlphaScreen secondary assay
3.3. Scaffold/Moiety Chemical Liabilities
The ML324 chemotype has a general drug-like nature and several marketed drugs possess quinoline scaffolds. In general, 8-hydroxy quinolones are possible metal chelators and could inhibit several metal dependent enzymes. However, these compounds are inactive against several metal dependent enzyme targets, such as 15-LOX and APE1 (data not shown). The probe molecule and several analogs obey Lipinski rule and showed excellent ADME properties. The probe does not contain any reactive functional groups or any known structural alerts. In addition, the molecule was stable in PBS buffer, assay buffer, mouse plasma and both in acidic and basic conditions between pH 2-10 over 48 hr. Overall, ML324 is a superior starting point with respect to potency and physiochemical properties for further development.
3.4. SAR Tables
Table 2Variations to compound 1
All Compounds synthesized at NCGC. IC50 values were measured using the AlphaScreen assay in triplicate.
 |
 |
 |
 |
 | |
|---|---|---|---|---|---|
| Entry | Internal ID | SID | CID | R | IC50 (μM) |
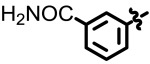
| 1 | NCGC00183784 | 104224071 | 459617 | NA | 0.84 |
| 2 | NCGC00183785 | 85736397 | 97394 | NA | 3.07 |
| 3 | NCGC00189502 | 104224401 | 49853311 |
 | 11.97 |
| 4 | NCGC00183808 ML324 | 85736407 | 44143209 |
| 0.92 |
| 5 | NCGC00183806 | 85736405 | 44143205 |
 | 1.62 |
| 6 | NCGC00183807 | 85736406 | 44143207 |
 | 4.27 |
| 7 | NCGC00244531 | 156647495 | 68740002 |
 | 1.24 |
| 8 | NCGC00247736 | 160646434 | 70679266 |
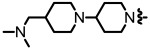 | 0.99 |
| 9 | NCGC00247763 | 160646435 | 70679265 |
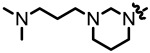 | 1.52 |
Table 3Variations to compound 1
All Compounds synthesized at NCGC. IC50 values were measured using the AlphaScreen assay in triplicate.
 | |||||
|---|---|---|---|---|---|
| Entry | Internal ID | SID | CID | R | IC50 (μM) |
| 10 | NCGC00241033 | 104224536 | 49852883 |
 | 2.25 |
| 11 | NCGC00241036 | 104224539 | 49852941 |
 | 1.74 |
| 12 | NCGC00241052 | 104224555 | 49852955 |
 | 1.33 |
| 13 | NCGC00241035 | 104224538 | 49852977 |
 | 1.57 |
| 14 | NCGC00189500 | 104224399 | 49852705 |
 | 2.24 |
| 15 | NCGC00241039 | 104224542 | 49852743 |
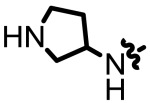 | 1.76 |
| 16 | NCGC00241043 | 104224546 | 49853042 |
 | 1.17 |
| 17 | NCGC00241048 | 104224551 | 49852972 |
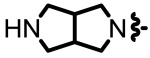 | 1.47 |
| 18 | NCGC00241045 | 104224548 | 49853264 |
 | 4.98 |
| 19 | NCGC00241754 | 104224686 | 49853290 |
 | 4.18 |
| 20 | NCGC00241753 | 104224685 | 49853053 |
 | 5.11 |
| 21 | NCGC00241759 | 104224691 | 49853381 |
 | 2.10 |
| 22 | NCGC00241756 | 104224688 | 49852707 |
 | 4.66 |
| 23 | NCGC00189508 | 104224407 | 49853351 |
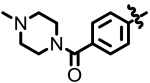
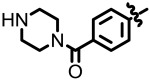 | 3.61 |
| 24 | NCGC00189504 | 104224403 | 49853247 |
 | 4.63 |
| 25 | NCGC00189503 | 104224402 | 49852863 |
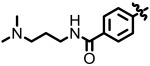 | 1.41 |
| 26 | NCGC00241765 | 104224698 | 49853324 |
 | 2.08 |
| 27 | NCGC00247752 | 160646433 | 70679264 |
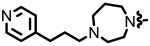 | 1.71 |
| 28 | NCGC00241055 | 104224558 | 49853387 |
 | 4.90 |
3.5. Cellular Activity
3.5.1. ML324 inhibits viral immediate early (IE) gene expression, reduced viral yields in HFF cells infected with HSV-1, and suppressed the formation of HSV plaques
%20Viral%20IE%20(ICP4%2C%20ICP27%2C%20ICP22)%20and%20control%20(S15%2C%20Sp1%2C%20TBP)%20mRNA%20levels%20in%20HFF%20cells%20treated%20with%20DMSO%20or%20the%20indicated%20concentrations%20of%20ML324%20for%203%20hrs%20and%20infected%20with%20HSV-1%20(0.&p=BOOKS&id=169450_ml324f14.jpg "Click on image to zoom")
Figure 11
(A) Viral IE (ICP4, ICP27, ICP22) and control (S15, Sp1, TBP) mRNA levels in HFF cells treated with DMSO or the indicated concentrations of ML324 for 3 hrs and infected with HSV-1 (0.1 pfu/cell) for 3 hr. (B) Viral yields from HFF cells treated with DMSO or ML324 for 3 hr and infected with HSV-1 (0.1 pfu/cell) for 24 hr in the presence of the drugs.
%20MRC-5%20cells%20were%20treated%20with%20DMSO%20or%20ML324%20at%20the%20indicated%20time%20relative%20to%20infection%20with%20HSV-1%20(1.&p=BOOKS&id=169450_ml324f15.jpg "Click on image to zoom")
Figure 12
(A) MRC-5 cells were treated with DMSO or ML324 at the indicated time relative to infection with HSV-1 (1.0 MOI). Viral and cellular mRNA levels were determined at 4 hr post infection. (B) Plaque assay of Vero cells infected with HSV-1 at the indicated MOI for 12 hr, followed by the addition of DMSO, ACV (100 μM), or ML324 (50 μM) for 48 hr. (C) Representative fields of MRC-5 and Vero cells infected with HSV-1 for 12 hr followed by addition of DMSO, ACV (100 μM) or ML324 (50 μM) for 48 hr (D) MRC-5 cells treated with DMSO or 50 μM ML324 were infected with HSV-1 at the indicated MOI. Viral IE and cellular mRNA levels were quantitated at 2 hr post infection; viral yields were determined at 24 hr post infection.
3.5.2. ML324 inhibits the spread of HSV infection to adjacent MRC-5 cells

Figure 13Immunofluorescent staining for the HSV DNA replication protein UL29
MRC-5 cells were infected with HSV-1 (0.1 MOI) for 8 hr, followed by the addition of DMSO, ACV (100 μM), or ML324 (50 μM) for 12 hr. Cells were stained with anti-UL29 (green), Dapi (blue), and Phalloidin-647 (F-actin, red).
3.5.3. ML324 exhibits activity in mouse sensory ganglia explant model
%20Viral%20yields%20from%20HSV-1%20latently%20infected%20trigeminal%20ganglia%20explanted%20in%20the%20presence%20of%20DMSO%2C%20or%202%20mM%20DMOG%20or%2050%20%003BCM%20ML324%20for%2048%20hr.&p=BOOKS&id=169450_ml324f17.jpg "Click on image to zoom")
Figure 14
(A) Viral yields from HSV-1 latently infected trigeminal ganglia explanted in the presence of DMSO, or 2 mM DMOG or 50 μM ML324 for 48 hr. [DMSO vs DMOG; Wilcoxon paired 2-tailed t test, p = .0015, n = 12; DMSO vs ML324, p < .0001, n = 20]. (B & D) Latently infected trigeminal ganglia were explanted in the presence of DMSO, 100 μM ACV, or 50 μM ML324 for 48 hr. Sections were costained with anti-UL29 (red), neurofilament 200 (green), and DAPI (blue) and scored for UL29(+) cell clusters (Clusters) and individual neurons (Single). [ACV vs ML324; 2-tailed t test, n = 48 sections representing 8 ganglia). The total number of UL29(+) per ganglia are graphed (D) and representative sections illustrated (B). Data shown are means +/- SEM. (C) Latently infected ganglia were explanted in the presence of DMSO, ACV (100 μM), or ML324 (50 μM) for 48 hr. Viral yields were determined from one half of each ganglia at 48 hr (DMSO, ACV, ML324) and from the other half of each ganglia after drug reversal for an additional 72 hr (ACV-R, ML324-R).
3.5.4. ML324 inhibits hCMV (IE) gene expression
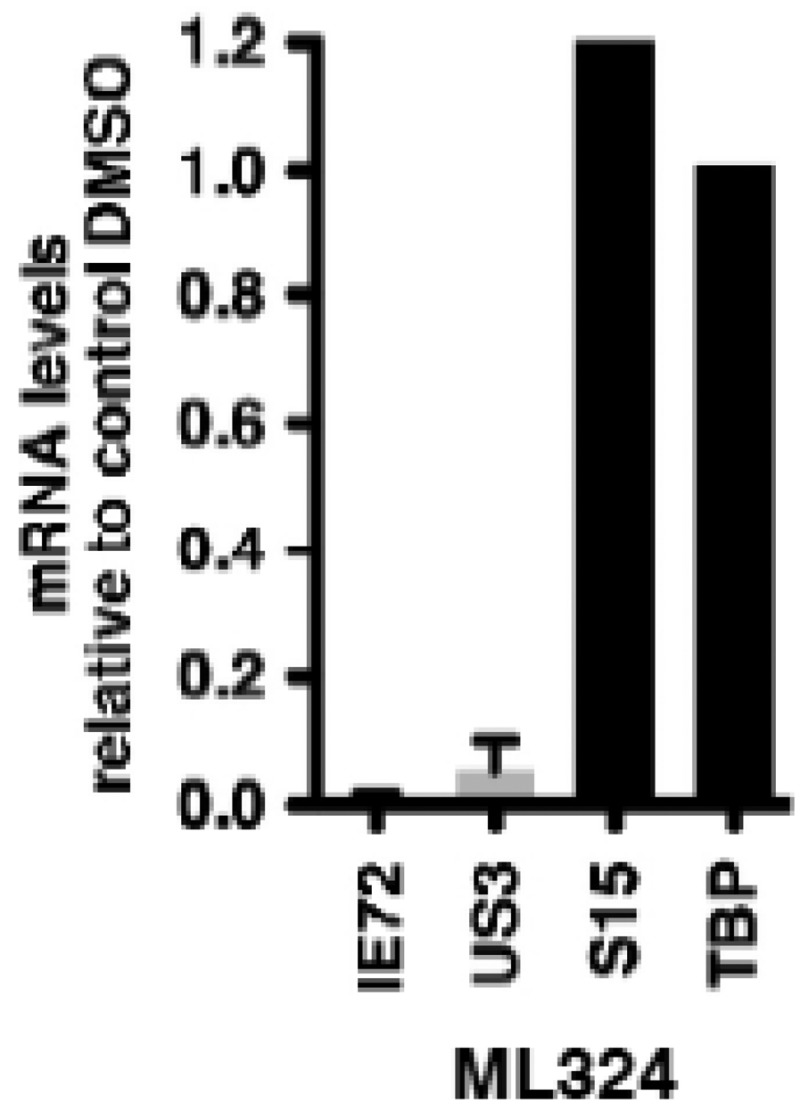
Figure 15MRC-5 cells were treated with DMSO or 50 mM ML324 for 3 hr, followed by infection with hCMV (0.1 pfu/cell) for 5 hr
mRNA levels of viral and cellular controls are expressed relative to cells treated with DMSO. Data shown are means +/- SEM.
3.6. Profiling Assays
3.6.1. ADME profile and in vivo PK for ML324
Table 4ADME profile for ML324
| Compound | PBS buffer (pH 7.4) Solubility (μM)a | Mouse Liver Microsome Stability (T1/2)a,b | Rat Liver Microsome Stability (T1/2)a,b | Caco-2 (A→B) Papp (10-6 cm/s)a | Caco-2 (B→A) Papp (10-6 cm/s)a | Efflux Ratio | Plasma Protein Binding (mouse)a | Mouse Plasma Stability (2 h)a |
|---|---|---|---|---|---|---|---|---|
| ML324 | 308 | >30 min | 72 min | 12.5 | 45.3 | 3.6 | 78% | 92% |
- a
Study was conducted by Pharmaron Inc. and was determined by LC/MS/MS
- b
represents the stability in the presence of NADPH. The probe compound showed no degradation without NADPH present over a 1 hr period.
4. Discussion
Structure Activity Relationships of ML324
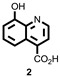
As previously reported, a high-throughput screening campaign of a roughly 230,000 compounds included in a diverse set of chemical library was carried out, using a formaldehyde dehydrogenase (FDH) assay, identified several 5-substituted-8-hydroxyquinolines as weak inhibitors of JMJD2E enzyme. [5] These compounds were further confirmed using an AlphaScreen assay platform which was ultimately used for subsequent structure activity (SAR) studies. 2,4-Pyridine dicarboxylic acid (2,4-PCDA) is a known non-selective inhibitor of JMJD2 enzymes with limited cell activity. On the basis of these observations and a overlaid model with the 8-HQ scaffold (identified in the HTS assay) and 2,4-Pyridine dicarboxylic acid, we designed and synthesized, 8-hydroxyquinoline-4-carboxylic acid (2) hoping to improve the potency and selectivity. Interestingly, despite the more obvious structural overlap with PDCA and compound 2, subsequent testing of 2 revealed the 8-hydroxyquinoline-5-carboxylic acid (1) to be more potent with IC50s of 3.1 and 0.84, respectively (Table 1). Successful co-crystal structure of 8-hydroxyquinoline-5-carboxylic acid (1) with JMD2A guided us to further explore around the 3-position of the quinoline core bearing a carboxylic acid functionality at 5-position (section 4.2, Figure 18). The quinoline nitrogen and the phenolic group were found to be essential to chelate the active site iron. SAR explorations of 3-position modifications were not easily accessible due to lack of literature precedence for this class of differentially substituted 8-HQs. As shown in Scheme 1, a novel and concise synthetic methodology was developed which allowed us to synthesize several analogs (10-28 as representative analogs, Table 3) via late-stage modifications at 3-position of the quinoline core bearing a carboxyilic acid at 5-position. The initial precursor 8-hydroxy-3-bromoquinoline was synthesized starting from commercially available 8-nitroquinoline using NBS-mediated bromination followed by iron reduction and heating the 8-amino-3-bromoquinoline with dilute sulfuric acid at 220 °C in a sealed tube. The phenolic group was protected with trimethylsilylethyl group using a Mitsunobu reaction to allow further chemical modifications of the key intermediate (Scheme 1). After extensive optimization of the reaction conditions, we were able to obtain the desired product in excellent yield via cross coupling of the t-butylcarbamate and the aryl bromide using a combination of palladium(II)acetate and tert-butyl XPhos catalyst system at 100 °C. Subsequent bromination of the formed product with NBS at low temperature provided predominantly the 7-brominated intermediate along with trace amount of dibrominated product. Palladium-catalyzed carbonylation using an alcohol (MeOH/EtOH) as a co-solvent afforded the 7-ester intermediate in good yield. Controlled deprotection of the t-butyl carbonyl group using trifluoroacetic acid in dichloromethane and subsequent Sandmeyer reaction of the amino group provided the aryl iodide. Suzuki or Buchwald cross coupling of this intermediate followed by the hydrolysis of the ester afforded varied types of analogs containing both hydrophobic and hydrophilic groups (selected analogs 10-28 are shown in the Table 3).
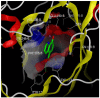
Figure 18
Co-crystallization of compound 1 with JMJD2A (PDB: 3NJY).
Of the few 3-substituted-quinolin-8-ol analogs without carboxylic acid functionality at 5-position synthesized (data not shown), all showed decreased potency against JMJD2E (example analog 3 with IC50 = 11.97 μM vs. analog 25 with IC50 = 1.41 μM). In general, the data suggested that the carboxylic acid was essential for the improved potency which was further supported by the co-crystal structure in which the carboxylic acid interacted with the tyrosine and lysine residues of the protein binding site. Introduction of hydrophilic groups at 3-position (analogs 10-28) improved the JMJD2E inhibition, whereas compounds with hydrophobic groups such as phenyl or methyl (data not shown) showed diminished activity. Based on these observations and the co-crystal structure, H-bond acceptor or donor moieties at 3-position seemed to facilitate a possible tight binding interaction with aspartate residue of the protein (data not shown). Unfortunately, as expected these analogs showed poor cell permeability due to the polar nature of the carboxylic acid functionality. Attempts to replace the carboxylic acid with tetrazole bioisostere or with other functional groups such as sulfonyl groups or the pro-drug approach with esters were met with limited success. In an attempt to address this problem, we envisioned optimization of the molecule at 6-position of the quinolin-8-ol without the carboxylic acid functionality. We postulated that the 5-COOH moiety anchored the 8-HQ into that particular orientation (3-substitutents towards the left for visualization purposes), whereas the des-carboxy analog may adopts a “flipped” binding mode. In this case, the 6-position of the HQ core structure would now occupy the space filled by the 3-substituted analogs described above (see Figure 16). As such, several 6-substituted quinolin-8-ol analogs (compounds 4-9 in Table 1 as representatives) were designed and prepared. Hydrophilic groups with H-bond donor or acceptor moieties in this region improved the potency against JMD2E enzyme (compounds 4-9 in Table 1). In contrast, hydrophobic groups were not well-tolerated in this region (data not shown). It is important to note that the compounds bearing amine tails attached to a para or meta benzamide linker (4-9) contributed to optimal potency with improved ADME properties. These observations might be attributed to interaction of the binding site via hydrogen bonding to nitrogen or oxygen atoms in this region. The summary of the SAR assessed during the course of the medicinal chemistry optimization is described below (Figure 17). In particular, compounds 4 (CID-44143209), 5 (CID-44143205), 7 (CID-68740002) 8 (CID-70679266) and 16 (CID-49853042) are highlighted as some of the potent candidates for further studies. However, we chose compound 4 (ML324) for further studies as it exhibited desired ADME properties and optimal activity in the HSV models for infection (vide infra).

Figure 16
Probe development/medicinal chemistry optimization flowchart.
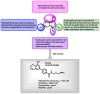
Figure 17
SAR summary of JMD2E probe, ML324.
In vitro ADME profile
One of the limitations with reported JMJD2 inhibitors is the modest potency and poor cell permeability. Thus, an important aspect of this program was to develop a novel inhibitor that possessed the required potency but also had favorable in vitro ADME properties (see section 3.6.1). ML324 was found to have an excellent solubility of 308 μM (PBS buffer) which is approaching the upper limit of the detection method. Typically, compounds with such favorable solubility have limited cell permeability as a result of their often polar nature. However, ML324 exhibits good Caco-2 cell permeability with a Papp of 12.5 (10-6 cm/s), albeit fairly significant efflux was observed, suggesting that it may be a Pgp substrate. These findings will need to be further explored by conducting the experiment in the presence of a Pgp inhibitor and see if the efflux is attenuated. We will also test permeability using the MDR1-MDCKII cell line, which also expresses Pgp to confirm the result. Importantly, ML324 possessed excellent microsomal stability in the presence of both mouse and rat liver microsomes with a T1/2 of >30 minutes and 72 minutes, respectively. Moreover, ML324 exhibited stability in plasma and aqueous solutions at various pH (see above) with low plasma protein binding of 78%. These data suggests that ML324 will have suitable pharmacokinetics/exposure for use in proof of concept animal models for HSV infection or studies requiring a JMJD2 inhibitor.
Biological Evaluation of ML324
Upon completion of the SAR studies of ML324, we had established that this compound has favorable potency, and in vitro ADME properties; we were eager to investigate the utility of ML324 in cell-based systems. Toward this end, we collaborated with an NIH colleague, Dr. Thomas Kristie (NIAID), who had previously demonstrated the use of histone demethylase LSD1 inhibitors as anti-HSV agents via suppression of viral lytic infection, accumulation of repressive chromatin on the viral IE gene regions, and repression of viral reactivation from latency. [4] Despite this promising activity, LSD1 is only responsible for H3K9 mono and di-methylation, but the JMJD2 family of demethylases is responsible for the removal of H3K9 trimethylation. As such, Kristie and co-workers sought to investigate the role of JMJD2 in HSV viral infection and the activation of IE gene expression via siRNA and pharmacological inhibition with ML324 and DMOG (di-methyloxaloglycine), a previously reported JMJD inhibitor. [14] Initial siRNA studies revealed that the JMJD2 family of histone demethylases is required for both HSV (herpes simplex virus) and hCMV (human cytomegalovirus) IE gene expression. [15] Moreover, depletion of JMJD2 suppressed the initiation of infection, blocked the spread of infection to adjacent cells, and resulted in suppression of reactivation in latently infected sensory neurons. Importantly, it was found that depletion of all four proteins (i.e.JMJD2A-D) was required to achieve maximal suppression of IE expression; depletion of individual JMJD2 members only had a modest impact. These data, along with the mechanistic redundancy of the JMJD2 family [16] suggested that a small molecule inhibitor selective for only one JMJD2 member may have limited efficacy in these models. We do not know the full selectivity of ML324 toward all JMJD2 family members, we have however found comparable activity between JMJD2A and JMJD2E which, given the structural similarity of these enzymes, is not surprising. Armed with this information, we sought to validate the siRNA findings through the use of ML324 in HSV and hCMV infection models.
The initial study tested the ability of ML324 to inhibit HSV viral IE infection in HFF cells. [14] As shown in Figure 11 (section: 3.5.1), ML324, potently reduced IE gene expression (IC50 = 10 μM) compared to 0.75 mM for DMOG, without an impact on the expression of the cellular controls Sp1, S15 and TBP. Next, HFF cells were treated with various concentrations of ML324 and infected with HSV-1 (0.1 pfu/cell) for 24 hr. ML324 reduced viral yields in a dose-dependent manner (∼4-5 logs at 25 μM, Figure 11 B) whereas 1.5 mM of DMOG was required to achieve the same reduction (data not shown). HSV-1 infected MRC-5 cells were treated with DMSO or ML324 at various time points (2 hr pre-infection to 4 hr post infection) and mRNA levels of IE viral genes (ICP4, ICP22, ICP27) were measured relative to DMSO at 4 hr post infection. As shown in Figure 12A, ML324, markedly reduced the gene transcription of all 4 viral IE genes, even when added 2 hr post-infection (>50% reduction). ML324 was tested for its ability to reduce viral yields in MRC-5 cells infected at high multiplicity of infection (MOI) (Figure 12 D), and was found to display comparable viral yield reduction at half the concentration needed for ACV (acyclovir), 50 μM vs. 100 μM respectively. We further demonstrated ML324 as an efficacious antiviral agent, by observing significant reduction in HSV plaque formation in Vero cells and MRC-5 with 50 μM, as compared to 100 μM ACV (Figure 12 B-C). Moreover, to investigate the effect of ML324 on the spread of infection to adjacent cells, MRC-5 cells were infected with HSV-1 and visualized by immunofluorescent staining for UL29 (HSV DNA replication protein) (see section 3.5.2, Figure 13). ML324 blocked viral gene expression at an early stage prior to expression of the viral replication protein UL29, thereby reducing the number of cells exhibiting viral antigens.
As demonstrated in section 3.5.3, Figure 14 A-D, ML324 exhibited significant antiviral activity in the mouse sensory ganglia explant model of HSV reactivation. First, ML324 reduced the viral yield per ganglia by 4.5 logs at 50 μM compared to DMOG which required concentrations up to 2 mM to achieve 1.5 log reduction (Figure 14 A). Immunofluorescent staining of explanted ganglia sections (Figure 14 B & D) revealed that ML324 was capable of suppressing the primary viral reactivation events. The spread of the reactivated viral infection of the ganglia was demonstrated in the DMSO control where the viral lytic gene (UL29) expression was observed in both isolated and clustered neurons (Figure 14 D). However, upon treatment with ML324, significant reduction in both isolated and clustered UL29(+) neurons was observed (note: ML324 was more efficacious than ACV in single neurons). Importantly, robust viral replication was observed following ML324 withdrawal which indicates the reduction in reactivation by ML324 was not simply due to the inability of the ganglia to support viral replication (Figure 14 C). In addition to the encouraging results obtained for the herpes simplex virus (HSV) reported herein, ML324 also demonstrated comparable activity (Section 3.5.4, Figure 15) towards β-herpes human Cytomegalovirus (hCMV) which causes mortality in immuno compromised patients and is the most significant cause for viral birth defects. [17] These data support the notion that the JMJD2 demethylases are required for both HSV and hCMV IE gene expression and demonstrate the effectiveness of ML324 as an antiviral agent.
4.1. Comparison to existing art and how the new probe is an improvement
All of the JMJD2 inhibitors reported to date suffer from poor cell permeability and modest potency with respect to JMJD2. Given the lack of cell permeability of the prior art, it has been difficult to study the role of JMJD2 demethylases in a cellular context using a small molecule inhibitor. This is exemplified by DMOG (dimethyloxalylgycline) used in the antiviral studies reported herein; ML324 exhibits cellular activity in the μM range, DMOG requires mM concentration to exert its effects. At such high compound concentrations it becomes difficult to attribute the observed biological effects to the engagement of studied target alone. In addition to the favorable cell permeability for the probe, ML324 also displays excellent microsomal stability, plasma stability, and lower protein plasma binding suggesting its use for in vivo studies. Further, ML324, is a drug-like small molecule which is amendable to medicinal chemistry optimization, whereas much of the prior art consists of amino-acid derived compounds which are likely to have limited bioavailability. GSK-1 represents the most potent and selective inhibitor of a JMJD family member to date (JMJD3), but it is also reported to have poor permeability and the corresponding pro-drug ester has modest cell activity. Moreover, this compound does not show activity towards the JMJD2 family and thus is not directly relevant as prior art for this family of enzymes.
4.2. Mechanism of action studies
Co-Crystallization of compound 1 (8-HQ-5-COOH)
Early co-crystallization efforts led to the successful crystallization of 1 to JMJD2A [5] which revealed that the 8-HQ scaffold is positioned in a similar location as the previously reported N-oxalylglycine analog described above. The quinoline-nitrogen and 8-hydroxy group coordinate with the active-site Ni(II) in a bidentate fashion. [Note: Ni(II) replaces Fe(II)] This structure also suggests that compound 1 competes with the binding of 2-OG but not directly with the peptide substrate. Though we do not yet have a co-crystal structure of the probe molecule ML324, the fact that a common 8-HQ core exists between the two suggests that they likely have comparable binding modes.
4.3. Planned Future Studies
The data presented above thoroughly establishes ML324 as a promising antiviral agent in cell-based models for HSV and hCMV (data not shown). In particular for HSV, ML324 was shown to suppress the initiation of infection, block the spread of infection to adjacent cells, and suppresses the reactivation of latently infected sensory neurons. A combination of siRNA studies and the use of ML324 support the notion that the JMJD2 family of histone demethylases is required for chromatin modulation and activation of IE transcriptiontranscription during HSV infection. Current studies involve the testing of these compounds in proof-of-concept animal models for HSV infection. Initial studies will consist of testing the compound as a single agent but future efforts will use ML324 in combination with LSD1 inhibitors which have already demonstrated in vivo efficacy in these models. Importantly, further work will be required to determine whether inhibition of these enzymes results in long-term suppression of the latent virus and the impact on subclinical shedding.
While the siRNA studies in HSV infection models suggest that a compound which possesses broad activity for the JMJD2 family (e.g. ML324) would be most optimal, it appears that a specific inhibitor for JMJD2D would be desirable for hCMV infection. [14] As such, we hope ML324 can be further optimized to possess more selectivity towards particular members of the JMJD2 family, perhaps via the aid of a co-crystal structure with JMJD2D. The excellent solubility and drug-like nature of ML324 should provide the basis for successful a crystallography trial and future medicinal chemistry efforts.
Separately, ML324 and related analogs have been requested by several collaborators around the world who are currently testing these compounds in cancer-related models and have already reported promising preliminary results (data not shown). We suspect that, given the favorable in vitro ADME properties of ML324 compared to previously reported inhibitors of this class of enzymes, the probe molecule and data included in this report will be of great interest to the scientific community.
5. References
- 1.
- Holliday R. Epigenetics: a historical review. 2006;1(2):76–80. [PubMed: 17998809]
- 2.
- Zhang Y, Reinberg D. Transcription regulation by histone methylation:interplay between different covalent modification of the core histone tails. Gene Dev. 2001;15(18):2343–2360. [PubMed: 11562345]
- 3.
- a. Bloom DC, Giordani NV, Kwiatkowski DL. Epigenetic modifications as therapeutic targets. Biochimica et Biophysica Acta. 2010;1799:246–256. [PMC free article: PMC2838971] [PubMed: 20045093]
b. Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nature Rev Microbiol. 2008;6:211–221. [PubMed: 18264117]
c. Giberson AN, Davidson AR, Parks RJ. Chromatin strucutre of adenovirus DNA throughout infection. Nucleic Acids Res. 2012;40:2369–2376. [PMC free article: PMC3315334] [PubMed: 22116065]
d. Kristie TM, Liang Y, Vogel JL. Control of alpa-herpes IE gene expression by HCF-1 coupled chromatin modification activities. Biochimica et Biophysica Acta. 2010;1799:257–265. [PMC free article: PMC2838944] [PubMed: 19682612] - 4.
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–1317. [PMC free article: PMC2783573] [PubMed: 19855399]
- 5.
- King ON, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, Klose RJ, Oppermann U, Jadhav A, Heightman TD, Maloney DJ, Schofield CJ, Simeonov A. Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors. PLoS One. 2010;5:e15535. [PMC free article: PMC2990756] [PubMed: 21124847]
- 6.
- Rose NR, Ng SS, Mecinović J, Liénard BM, Bello SH, Sun Z, McDonough MA, Oppermann U, Schofield CJ. Inhibitor scaffolds for 2-oxoglutarate-dependent histone lysine demethylases. J. Med. Chem. 2008;51:7053–7056. [PubMed: 18942826]
- 7.
- Hamada S, Kim TD, Suzuki T, Itoh Y, Tsumoto H, Nakagawa H, Janknecht R, Miyata N. Synthesis and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg Med Chem Lett. 2009;19:2852–2855. [PubMed: 19359167]
- 8.
- Thalhammer A, Mecinović J, Loenarz C, Tumber A, Rose NR, Heightman TD, Schofield CJ. Inhibition of the histone demethylase JMJD2E by 3-substituted pyridine 2,4-dicarboxylates. Org Biomol Chem. 2011;9:127–135. [PMC free article: PMC4733660] [PubMed: 21076780]
- 9.
- Hamada S, Suzuki T, Mino K, Koseki K, Oehme F, Flamme I, Ozasa H, Itoh Y, Ogasawara D, Komaarashi H, Kato A, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Design, synthesis, enzyme-inhibitory activity, and effect on human cancer cells of a novel series of jumonji domain-containing protein 2 histone demethylase inhibitors. J Med Chem. 2010;53:5629–5638. [PubMed: 20684604]
- 10.
- Woon E.C, Tumber A, Kawamura A, Hillringhaus L, Ge W, Rose NR, Ma JH, Chan MC, Walport LJ, Che KH, Ng SS, Marsden BD, Oppermann U, McDonough MA, Schofield CJ. Linking of 2-oxoglutarate and substrate binding sites enables potent and highly selective inhibition of JmjC histone demethylases. Angew Chem Int Ed Engl. 2012;51:1631–1634. [PubMed: 22241642]
- 11.
- Rose NR, Woon EC, Kingham GL, King ON, Mecinović J, Clifton IJ, Ng SS, Talib-Hardy J, Oppermann U, McDonough MA, Schofield CJ. Selective inhibitors of the JMJD2 histone demethylases: combined nondenaturing mass spectrometric screening and crystallographic approaches. J Med Chem. 2010;53:1810–1818. [PMC free article: PMC2825117] [PubMed: 20088513]
- 12.
- Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. [PMC free article: PMC4691848] [PubMed: 22842901]
- 13.
- Sakurai M, Rose NR, Schultz L, Quinn AM, Jadhav A, et al. A miniaturized screen for inhibitors of Jumonji histone demethylases. Mol Biosyst. 2010;6:357–364. [PMC free article: PMC3401096] [PubMed: 20094655]
- 14.
- Hamada S, Kim TD, Suzuki T, Itoh Y, Tsumoto H, Nakagawa H, Janknecht R, Miyata N. Syntheiss and activity of N-oxalylglycine and its derivatives as Jumonji C-domain-containing histone lysine demethylase inhibitors. Bioorg. Med. Chem. Lett. 2009;19:2852–2855. [PubMed: 19359167]
- 15.
- Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM. Targeting JMJD2 histone demethylases to epigenetically control herpes infection and reactivation from latency. Sci. Transl. Med. accepted. [PMC free article: PMC4778723] [PubMed: 23303604]
- 16.
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of Histone Lysine Trimethylation by the JMJD2 Family of Histone Demethylases. 2006;125:467–481. [PubMed: 16603238]
- 17.
- a. Britt W. Human Herpesviruses Biology, Therapy, and Immunoprophylaxis. Arvin A, Whitley R, editors. Cambridge University Press; Cambridge: 2007. pp. 737–764. [PubMed: 21348071]
b. Mocarski ES, Shenk T, Pass RF. Fields Virology. Fifth Edition.
b. Knipe DM, Howley PM, editors. Vol. 2. Lippincott Williams & Wilkins; Philapelphia: 2007. pp. 2701–2772.
6. Appendix
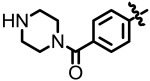
(4-(8-Hydroxyquinolin-6-yl)phenyl)(piperazin-1-yl)methan-one (NCGC00183806)

LC-MS Retention Time: t1 (Method 1) = 2.812 min and t2 (Method 2) = 2.359 min; 1H NMR (400 MHz, DMSO-d6) d 3.09 – 3.2 (m, 4 H) 3.52 - 4.58 (m, 4 H) 7.47 (d, J = 1.4 Hz, 1 H) 7.57 - 7.71 (m, 3 H) 7.81 (s, 1 H) 7.89 (d, J = 8.0 Hz, 2 H) 8.49 (d, J = 8.2 Hz, 1 H) 8.89 – 8.92 (m, 3 H); HRMS (ESI) m/z (M+H)+ calcd. for C20H20N3O2, 334.1550; found 334.1555.
3-(4-Aminopiperidin-1-yl)-8-hydroxyquinoline-5-carboxylic acid (NCGC00241035)

LC-MS Retention Time: t1 (Method 1) = 2.645 min and t2 (Method 2) = 2.268 min; 1H NMR (400 MHz, DMSO-d6) d ppm 1.61 - 1.78 (m, 2 H) 2.03 - 2.05 (m, 2 H) 2.97 – 2.99 (m, 3 H) 3.67 – 3.97 (m, 4 H) 6.88 (d, J = 8.4 Hz, 1 H) 7.90 (m, 2 H) 8.19 (d, J = 8.2 Hz, 2 H) 8.80 (d, J = 2.7 Hz, 1 H) 8.85 (d, J =2.7 Hz, 2 H); HRMS (ESI) m/z (M+H)+ calcd. for C15H18N3O3, 288.1343; found 288.1342.
(3-(8-Hydroxyquinolin-6-yl)phenyl)(4-methylpiperazin-1-yl)methanone (NCGC00183807)

LC-MS Retention Time: t1 (Method 1) = 2.931 min and t2 (Method 2) = 2.393 min; 1H NMR (400 MHz, DMSO-d6) d ppm 2.81 (s, 3 H) 2.97 - 3.62 (m, 8 H) 7.42 - 7.49 (m, 2 H) 7.55 - 7.70 (m, 2 H) 7.79 (d, J = 12.9 Hz, 2 H) 7.89 (d, J = 7.8 Hz, 1 H) 8.48 (d, J = 8.2 Hz, 1 H) 8.87 (d, J = 3.3 Hz, 1 H) 10.01 (br. s., 1 H); HRMS (ESI) m/z (M+H)+ calcd. for C21H22N3O2, 348.1707; found 348.1695.
3-(8-Hydroxyquinolin-6-yl)-N-(3-(piperazin-1-yl)- propyl)benzamide (NCGC00244531)

LC-MS Retention Time: t1 (Method 1) = 2.758 min and t2 (Method 2) = 2.380 min; 1H NMR (400 MHz, DMSO-d6) d ppm 1.87 - 1.95 (m, 2H), 3.47 – 3.02 (m, 12H), 7.52 (d, J = 1.9 Hz, 1H), 7.71 – 7.58 (m, 2H), 7.81 (d, J = 1.9 Hz, 1H), 7.89 (ddd, J = 7.8 Hz, 1.7 Hz and 1.1 Hz, 1H), 7.96 (ddd, J = 7.7 Hz, 1.9 Hz and 1.1 Hz, 1H), 8.25 (t, J = 1.7 Hz, 1H), 8.47 (dd, J = 8.4 Hz and 1.6 Hz, 1H), 8.89 (dd, J = 4.3 Hz and 1.6 Hz, 1H), 8.78 (t, J = 5.7 Hz, 1H); HRMS (ESI) m/z (M+H)+ calcd. for C23H27N4O2, 391.2129; found 391.2131.
N-(3-(dimethylamino)propyl)-4-(8-hydroxyquinolin-3-yl)benzamide (NCGC00189502)

LC-MS Retention Time: t1 (Method 1) = 2.945 min and t2 (Method 2) = 2.471 min; 1H NMR (400 MHz, DMSO-d6) d ppm 1.84 - 1.99 (m, 2 H) 2.80 (s, 6 H) 3.08 - 3.19 (m, 2 H) 3.35 – 3.339 (m, 2 H) 7.15 (t, J = 4.4 Hz, 1 H) 7.52 (d, J = 4.3 Hz, 2 H) 8.01 – 8.05 (m, 4 H) 8.70 – 8.75 (m, 2 H) 9.23 (d, J = 2.2 Hz, 1 H) 9.51 (br. s., 1 H); HRMS (ESI) m/z (M+H)+ calcd. for C21H24N3O2, 350.1863; found 350.1874.

Figure S1LC/MS trace of ML324
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency.[mBio. 2013]A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency.Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM. mBio. 2013 Feb 5; 4(1):e00558-12. Epub 2013 Feb 5.
- Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency.[Sci Transl Med. 2013]Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency.Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM. Sci Transl Med. 2013 Jan 9; 5(167):167ra5.
- Histone Lysine Demethylases of JMJD2 or KDM4 Family are Important Epigenetic Regulators in Reward Circuitry in the Etiopathology of Depression.[Neuropsychopharmacology. 2017]Histone Lysine Demethylases of JMJD2 or KDM4 Family are Important Epigenetic Regulators in Reward Circuitry in the Etiopathology of Depression.Pathak SS, Maitra S, Chakravarty S, Kumar A. Neuropsychopharmacology. 2017 Mar; 42(4):854-863. Epub 2016 Oct 6.
- Review LSD1 and the chemistry of histone demethylation.[Curr Opin Chem Biol. 2007]Review LSD1 and the chemistry of histone demethylation.Culhane JC, Cole PA. Curr Opin Chem Biol. 2007 Oct; 11(5):561-8. Epub 2007 Sep 11.
- Review The future therapeutic potential of histone demethylases: A critical analysis.[Curr Opin Drug Discov Devel. 2...]Review The future therapeutic potential of histone demethylases: A critical analysis.Natoli G, Testa G, De Santa F. Curr Opin Drug Discov Devel. 2009 Sep; 12(5):607-15.
- Discovery of ML324, a JMJD2 demethylase inhibitor with demonstrated antiviral ac...Discovery of ML324, a JMJD2 demethylase inhibitor with demonstrated antiviral activity - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...