

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

A Next generation PLD2 inhibitor with improved physiochemical properties and DMPK profile for translational in vivo
Matthew C. O'Reilly, Sarah A. Scott, J. Scott Daniels, Ryan Morrison, Julie L. Engers, Thomas Oguin, Paul Thomas, H. Alex Brown, and Craig W. Lindsley.
Author Information and Affiliations .
.Received: April 15, 2014; Last Update: January 16, 2015.
Further chemical optimization of our first generation phospholipase D2 (PLD2) probe (ML298) afforded ML395 (CID 73099363), a potent, >80-fold PLD2 selective allosteric inhibitor (cellular PLD1, IC50 >30,000 nM, cellular PLD2, IC50 = 360 nM). ML395 displays no inhibition of PLD1, displays exceptional solubility, stability and PK; therefore, ML395 is an improvement over our first generation PLD2 selective inhibitor, ML298. ML395 possesses favorable physiochemical and dystrophia myotonica-protein kinase (DMPK) properties, making it a useful tool to probe selective PLD2 function in vitro and in vivo. Data here shows therapeutic relevance in virology with no observed toxicity at very high doses.
Assigned Assay Grant #: P01 ES013125
Screening Center Name & PI: Vanderbilt Specialized Chemistry Center for Accelerated Probe Development, Craig Lindsley (H. Alex Brown – Med Chem Fast Track); St. Jude's Children's Research Hospital
Chemistry Center Name & PI: Vanderbilt Specialized Chemistry Center for Accelerated Probe Development, Craig Lindsley
Assay Submitter & Institution: H. Alex Brown, Vanderbilt University
PubChem Summary Bioassay Identifier (AID): 588854
Probe Structure & Characteristics

ML395
| CID/ML# | Target Name | IC50/(nM) [SID, AID] | Anti-target Name(s) | IC50 (μM) [SID, AID] | Fold Selective | Secondary Assay(s) Name: IC50/EC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 73099363/ML395 | PLD2 (cellular) | 360 nM [SID 173128374, AID 743409, AID 743400] | PLD1 (cellular) | 30,000 nM [SID 173128374, AID 743388, AID 743389] | >80 | Biochemical Assay PLD1 = >30,000 nM, PLD2 = 8700 nM [SID 173128374, AID 743390, AID 743391] |
1. Recommendations for scientific use of the probe
ML395 (CID 73099363) is a potent, >80-fold PLD2 selective allosteric inhibitor (cellular PLD1, IC50 >30,000 nM, cellular PLD2, IC50 = 360 nM). ML395 can be used both in vitro and in vivo to study the role of selective inhibition of phospholipase D2 (PLD2) in a manner previously unavailable, as our first generation inhibitors (ML298 and ML299) possessed only modest PK, modest solubility and toxicity at higher concentrations. Moreover, ML395 possess an attractive in vitro and in vivo DMPK profile, ancillary pharmacology (Eurofins Panlabs Panel) cleaner than any toher reported PLD inhibitor, and has been found to possess interesting activity as an antiviral agent in cellular assays.
Introduction
Phospholipase D (PLD) is a lipid signaling enzyme that catalyzes the hydrolysis of phosphatidylcholine (PC, 1) into the lipid second messenger phosphatidic acid (PA, 2) and choline 3 (Figure 1A).1-3 PA is an important lipid second messenger that is strategically located at the intersection of several critical signaling and metabolic pathways. Two mammalian isoforms of PLD have been identified, designated PLD1 and PLD2 (Figure 1B), which share 53% sequence identity and are subject to different regulatory mechanisms. Both isoforms share a conserved histidine, lysine, aspartate (HKD) amino acid domain that forms the catalytic site, as well as conserved phox homology (PX) and pleckstrin homology (PH) regulatory domains at the N-terminus.1-3 Dysregulated PLD function has been implicated in, and genetic and biochemical experiments indicate a role for PLD in, cancer (breast, renal, colorectal and glioblastoma),4-7 CNS disorders (such as Alzheimer's disease8 and stroke9) as well as in key stages of viral infection.10 However, the tools available to inhibit PLD activity have been limited to genetic and biochemical approaches including the use of n-butanol, none of which are viable therapeutic options.1
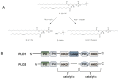
Furthermore, n-butanol is not a PLD inhibitor, rather n-butanol blocks PLD-catalyzed phosphatidic acid production by competing with water as a nucleophile thereby causing the formation of phosphatidylbutanol 4 in a competitive transphosphatidylation reaction. Developing isoform-selective PLD inhibitors is a formidable task due to several factors: (1) difficulty in producing and purifying PLD1 and PLD2 on scale, (2) labor intensive and time consuming enzymatic assays, and (3) high sequence homology between PLD1 and PLD2. Importantly, due to the multitude of cellular events which require PA, ablating all PLD enzymatic activity may not be a viable therapeutic approach, in which case it is necessary to develop isoform-selective PLD inhibitors.
Prior Art. Unfortunately, there have been very few small molecule tools to study PLD function and, no small molecule tool existed to dissect the individual roles of PLD1 and PLD2. Historically, the field relied on the over-expression of either catalytically active or inactive forms of either PLD1 or PD2 in vivo, or employed siRNA for the individual isoforms in an effort to discern discrete roles for PLD1 and PLD2.1 In order to assess the therapeutic potential of the inhibition of PLD1, PLD2 and/or dual inhibition of both isoforms, the genetic and biological data must be recapitulated with a small molecule. Sadly, small molecule PLD inhibitors with the profile to serve as probes were lacking, and none of the early inhibitors afforded isoform selective inhibition.1 Moreover, the most utilized small molecule to study PLD function over the past 20 years is n-butanol, a compound inaccurately described in the literature, as recently as 2011, as a PLD inhibitor (Figure 2). It is important to emphasize that n-butanol is not a PLD inhibitor, rather n-butanol (as well as other primary alcohols) block PLD-catalyzed phosphatidic acid (PA) production by competing with water as a nucleophile thereby causing the formation of phosphatidylbutanol in a transphosphatidylation reaction. In addition, there are concerns that n-butanol may not fully block PA production and n-butanol may also be promiscuous in cell-based assays affecting multiple pathways in addition to transphosphatidylation. Thus, conclusions reached in the literature from studies employing n-butanol alone should be viewed with caution, and the data generated require further confirmation with the next generation of isoform selective small molecule PLD inhibitors. Over the past twenty years, a diverse range of chemotypes 5-2111-40 have been reported as inhibitors of either PLD or PLD signaling (Figure 2) based on activity in an equally diverse array of dissimilar PLD assays; thus, quantitative, and in some instances qualitative, comparisons with regards to PLD activity are not possible. As a result, early PLD inhibitors fall into two categories: indirect inhibitors (compounds that inhibit PLD enzymatic activity in cells and/or decrease PLD protein expression in cells, but do not directly inhibit PLD enzymatic activity in vitro) and direct inhibitors (compounds that decrease PLD enzymatic activity measured by transphosphatidylation in cells and measured by the hydrolysis of 3H-phosphatidylcholine in an in vitro reconstitution assay). As many of these inhibitors have not been thoroughly studied, these divisions by mechanism of action should also be taken with caution, and in several cases, a label of mechanism unknown may be more appropriate. Overall, the prior art is comprised of natural products, phosphonate mimetics and synthetically-derived small molecules.1,11-40

In 2007, the PLD filed was energized by a brief report from a group at Novartis41 on a high throughput screen to identify PLD2 inhibitors, which led to the identification of halopemide (22), a psychotropic agent originally reported by Janssen42 in the early 1980s for numerous neuroscience indications as a PLD2 inhibitor with an IC50 of 1.5 μM (Figure 3).41 However, there was no mention of PLD1 inhibition in this initial short paper (or assay details), but it was subsequently found that halopemide 22, in our lab,2 potently inhibits both PLD1 (cellular IC50 = 21 nM, biochemical IC50 = 220 nM) and PLD2 (cellular IC50 = 300 nM, biochemical IC50 = 310 nM) Thus, halopemide (22) is more accurately described as a dual PLD1/2 inhibitor, and actually shows a preference for PLD1 inhibition. Halopemide (22), also known as R 34301, is related to the butyrphenone-based neuroleptics, such as haloperidol, which was originally developed as an anti-emetic drug, but later found to possess unique psychotropic effects as a dopamine antagonist (D2 IC50 = 7 nM).42 22 was found to be a ‘psychic energizer’ having effects on the negative symptoms, as well as the positive symptoms, of schizophrenia without the extrapyramidal side effects common to standard atypical antipsychotic agents. Halopemide, like most atypical antipsychotics, is an example of polypharmacology (highly promiscuous pharmacology, inhibiting and activating a number of biogenic amine receptors in the CNS). Halopemide was evaluated in five separate clinical trials with over 100 schizophrenic, oligophrenic and autistic patients receiving the drug, and efficacy was observed in the majority of patients, and importantly, no adverse side effects or toxicities were noted, despite achieving plasma exposures of 100 ng/mL to 360 ng/mL from the 20 mg/kg and 60 mg/kg doses of 22, respectively.42 At these plasma concentrations, PLD1 was clearly inhibited, suggesting inhibition of PLD by this chemotype is safe in humans and a therapeutically viable mechanism. Thus, the halopemide (22) scaffold is an excellent starting point upon which to base a PLD inhibitor development campaign due to the potent PLD inhibition, favorable preclinical DMPK profile, and most importantly, extensive history in multiple clinical trials. The challenges are to get outside patented chemical space, development of both dual PLD1/2 and isoform selective PLD inhibitors (Both PLD1 and PLD2) and dial-out the D2 and other biogenic amine activity in the parent 22. A diversity-oriented synthesis (DOS) approach generated ∼260 analogs, from which VU0359595 (23) emerged as a 1,700-fold PLD1 selective inhibitor. The benzimidazolone core always favored PLD1 inhibition. Also from this effort, we identified a triazaspirone scaffold that could replace the benzimidizolone, but afforded, for the first time, preferential PLD2 inhibition. The first PLD2 preferring compound was VU0285655 (24), displaying ∼21-fold preference for inhibition of PLD2 over PLD1. A 4 × 6 matrix library, based on 4 acid chlorides and 6 halogenated triazaspirone cores, identified a 3-F phenyl triazaspirone and a 2-naphthyl amide as the best combination of elements to afford an improved PLD2 inhibitor, ie, VU0364739 (25), that displays 75-fold preference for inhibition of PLD2 over PLD1. However, 25 still affords potent inhibition of PLD1 (IC50 = 1,500 nM), and at standard 10 μM in vitro studies and in vivo plasma exposures, both PLD1 and PLD2 will be inhibited. Thus, there was significant room for improvement. Moreover, only 4 amides were surveyed in the context of the more optimal 1-(3-fluorophenyl)-1,3,8-triazspiro[4.5]decan-4-one 26, and future work would evaluate additional amides. Furthermore, the benzimidazolone moiety engendered more ancillary pharmacology at biogeneic amines than the triazaspirone core, and thus represents a more attractive starting point for further optimization of both PLD1 and PLD2 inhibitors.1,2,43,44 Subsequent optimization efforts led to the development of ML298 (26), an ∼75-fold selective PLD2 inhibitor as well as ML299 (27) a potent dual PLD1/PLD2 inhibitor, both with improved ancillary pharmacology and DMPK relative to 22-25. However, despite these advances, DMPK was still moderate, solubility was modest and at higher concentrations, toxicity was noted. Thus, the optimal in vivo probes to dissect PLD function still remained elusive.
2. Materials and Methods
Cell Culture.2,3 Calu-1, and MDA-231 cells were purchased from American Type Culture Collection (Manassas, VA). The 4T1 cell line was a gift from F. Miller (Karmanos Cancer Center, Detroit, MI). The PMT cells were derived from mammary tumors arising in the MMTV/PyVmT transgenic mice. Calu-1, PMT and 4T1 cells were maintained in DMEM supplemented with 10% FBS, 100 μg/mL penicillin-streptomycin and 0.25 μg/mL amphotericin. MDA-MB-231 cells were grown in Improved MEM supplemented with 10% FBS, 100 μg/mL penicillin-streptomycin and 0.25 μg/mL amphotericin. HEK293 cells stably expressing GFP tagged human PLD2A were generated in the lab. To sustain selection pressure low passage-number HEK293-gfpPLD2 cells were maintained in DMEM supplemented with 10% FBS, 100 μg/mL penicillin-streptomycin, 2 μg/mL puromycin and 600 μg/mL G418. Cells were used only for a limited number of passages because after ∼8 or 9 passages cells became generally unhealthy and growth rate and viability was greatly diminished. All HEK293-gfpPLD2 experiments were done on tissue culture plates that had been coated with poly-lysine (1 μg per well for 12-well plates or 2 μg per well for 6-well plates). All cells were maintained in a humidified 5% CO2 incubator at 37 °C.
In vitro measurement of PLD activity (Biochemical Assay).2,3 In vitro PLD activity was measured with an exogenous substrate assay as previously described (Henage et al. 2006, Brown et al. MIE 2007). Briefly, PLD activity was measured as the release of [methyl-3H] choline from [choline-methyl-3H] dipalmitoyl-phosphatidylcholine. 3–50 nM PLD was reconstituted with phospholipid vesicle substrates composed of 10 μM dipalmitoyl-PC, 100 μM PE (bovine liver), 6.2 μM PIP2 (porcine brain), and 1.4 μM cholesterol. Lipid solutions were dried under a gentle stream of nitrogen and resuspended in 100 mM Hepes, pH 7.5, 160 mM KCl, 6 mM EGTA, 0.2 mM DTT. Small unilamellar vesicles were prepared by bath sonication (2× 2-min intervals at 80 watts). All assays were performed at 37 °C on agitation for 30 minutes in 50 mM Hepes, pH 7.5, 80 mM KCl, 3 mM EGTA, 0.1 mM DTT, 3.6 mM MgCl2, 3.6 mM CaCl2, and 10 μM GTPγS. Reactions were stopped with the addition of trichloroacetic acid and bovine serum albumin. Free [methyl-3H] choline was separated from precipitated lipids and proteins by centrifugation, and was analyzed by liquid scintillation counting. Raw data is normalized in order to subtract background radioactivity levels, and is presented as percent total activity. All experiments were performed in triplicate.
Endogenous (Cellular) PLD activity assay of cell lines using deuterated 1-butanol incorporation.2,3 Endogenous PLD activity was determined using a modified in vivo deuterated 1-butanol PLD assay. Cells were seeded into 12-well tissue culture plates to reach 100% confluence at time of assay. All cell types aside from the HEK293-gfpPLD2 cells were serum starved 18 hours prior to experiment in DMEM, 0.5% FBS, 1%AA. Cells were pretreated in the presence of PLD inhibitor (ranging from 2 μM to 200 pM final concentration) or DMSO (vehicle control) in DMEM for 5 minutes at room temperature. After pretreatment media was removed, cancer cells were treated with DMEM + 1 μM PMA + 0.3% 1-butanol-d10 and either PLD inhibitor or DMSO vehicle control, or DMEM alone for 30 minutes at 37 ºC. HEK293-gfpPLD2 cells were treated in the presence of DMEM + 0.3% 1-butanol-d10 and either PLD inhibitor or DMSO but were not stimulated with PMA in any of the conditions. After treatment samples were extracted and internal standard was added. The resulting lipids were dried and was resuspended in MS solvent (9:1 methanol:chloroform + 1 μL NH4OH). Samples were then directly injected into a Finnigan TSQ Quantum triple quadrupole mass spectrometer and data was collected in negative ionization mode. Data was analyzed by plotting the ratio of the major phosphatidylbutanol-d9 lipid products of PLD stimulation divided by 32:0 PtdMeOH ((34:1 PtdBuOH (m/z 738) + 32:1 PtdBuOH(m/z 710))/PtdMeOH internal standard (m/z 661)). Background signal was subtracted using cells not treated with 1-butanol-d10 as a negative control. The data was then compared to vehicle control samples and is expressed as % of PMA stimulated PLD activity as in the case of the various cancer cell lines or as % of basal PLD activity.
DMPK Methods. In vitro: The metabolism of ML395 was investigated in rat hepatic microsomes (BD Biosciences, Billerica, MA) using substrate depletion methodology (% test article remaining). A potassium phosphate-buffered reaction mixture (0.1 M, pH 7.4) of test article (1 μM) and microsomes (0.5 mg/mL) was pre-incubated (5 min) at 37°C prior to the addition of NADPH (1 mM). The incubations, performed in 96-well plates, were continued at 37 °C under ambient oxygenation and aliquots (80 μL) were removed at selected time intervals (0, 3, 7, 15, 25 and 45 min). Protein was precipitated by the addition of chilled acetonitrile (160 μL), containing glyburide as an internal standard (50 ng/mL), and centrifuged at 3000 rpm (4°C) for 10 min. Resulting supernatants were transferred to new 96-well plates in preparation for LC/MS/MS analysis. The in vitro half-life (t1/2, min, Eq. 1), intrinsic clearance (CLint, mL/min/kg, Eq. 2) and subsequent predicted hepatic clearance (CLhep, mL/min/kg, Eq. 3) were determined employing the following equations:
Plasma Protein Binding. Protein binding of ML395 was determined in rat plasma via equilibrium dialysis employing Single-Use RED Plates with inserts (ThermoFisher Scientific, Rochester, NY). Briefly plasma (220 μL) was added to the 96 well plate containing test article (5 μL) and mixed thoroughly. Subsequently, 200 μL of the plasma-test article mixture was transferred to the cis chamber (red) of the RED plate, with an accompanying 350 μL of phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated 4 h at 37 °C with shaking. At completion, 50 μL aliquots from each chamber were diluted 1:1 (50 μL) with either plasma (cis) or buffer (trans) and transferred to a new 96 well plate, at which time ice-cold acetonitrile (2 volumes) was added to extract the matrices. The plate was centrifuged (3000 rpm, 10 min) and supernatants transferred to a new 96 well plate. The sealed plate was stored at -20 °C until LC/MS/MS analysis.
Liquid Chromatography/Mass Spectrometry Analysis. In vitro experiments. ML298 was analyzed via electrospray ionization (ESI) on an AB Sciex API-4000 (Foster City, CA) triple-quadrupole instrument that was coupled with Shimadzu LC-10AD pumps (Columbia, MD) and a Leap Technologies CTC PAL auto-sampler (Carrboro, NC). Analytes were separated by gradient elution using a Fortis C18 2.1 × 50 mm, 3.5 μm column (Fortis Technologies Ltd, Cheshire, UK) thermostated at 40 °C. HPLC mobile phase A was 0.1% NH4OH (pH unadjusted), mobile phase B was acetonitrile. The gradient started at 30% B after a 0.2 min hold and was linearly increased to 90% B over 0.8 min; held at 90% B for 0.5 min and returned to 30% B in 0.1 min followed by a re-equilibration (0.9 min). The total run time was 2.5 min and the HPLC flow rate was 0.5 mL/min. The source temperature was set at 500°C and mass spectral analyses were performed using multiple reaction monitoring (MRM) utilizing a Turbo-Ionspray® source in positive ionization mode (5.0 kV spray voltage). LC/MS/MS analysis was performed employing a TSQ QuantumULTRA that was coupled to a ThermoSurveyor LC system (Thermoelectron Corp., San Jose, CA) and a Leap Technologies CTC PAL auto-sampler (Carrboro, NC). Chromatographic separation of analytes was achieved with an Acquity BEH C18 2.1 × 50 mm, 1.7 μm column (Waters, Taunton, MA).
2.1. Assays
- 2.1.1.
Summary AID 588854: PLD_CRC (Summary)
- 2.1.2.1.
AID 743388: Identifying inhibitors with PLD1 specific Cellular Isoenzyme Activity
- 2.1.2.2.
AID 743389: PLD1 Cellular Concentration Response Study
- 2.1.2.3.
AID 743390: PLD1 Inhibitor Confirmatory assay using the Purified Enzyme Concentration Response Method
- 2.1.2.4.
AID 743391: Discovery of PLD2 inhibitors using the Purified Enzyme Concentration Response Assay
- 2.1.2.5.
AID 743400: PLD2 Cellular Concentration Response Study
- 2.1.2.6.
AID 743409: Identifying inhibitors with PLD2 specific Cellular Isoenzyme Activity
- 2.1.2.7.
AID 743437: Development of inhibitors for PLD2 (Eurofin Panel Assay Results)
2.2. Probe Chemical Characterization

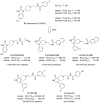
Probe compound ML395 (CID 73099363, SID 173128374) was prepared according to the above scheme and had the following characterization. N-(2-(4-oxo-1-(pyridine-3-ylmethyl)-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)2-naphthamide: 1H NMR (400.1 MHz, MeOD) δ (ppm): 8.53-8.36 (m, 3H); 8.01-7.72 (m, 6H); 7.56-7.47 (m, 2H); 7.34 (dd, J1 = 7.7 Hz, J2 = 4.9 Hz, 1H); 3.88 (s, 2H); 3.72 (s, 2H); 3.61 (t, J = 6.8 Hz, 2H); 3.35 (s, 1H); 2.92-2.79 (m, 4H); 2.69 (t, J = 6.8 Hz, 2H); 1.89 (td, J1 = 13.5 Hz, J2 = 5.0 Hz, 2H); 1.82-1.73 (m, 2H). 13C NMR (100.6 MHz, MeOD) δ (ppm): 179.96, 169.99, 150.08, 149.00, 138.37, 136.23, 136.14, 133.96, 132.81, 129.97, 129.29, 128.79, 128.77, 128.74, 127.81, 125.19, 124.87, 62.19, 60.88, 58.03, 50.48, 48.85, 38.34, 29.66. HRMS (TOF, ES+) C26H30N5O2 [M+H]+ calc. mass 444.2400, found 444.2402.
Solubility. Solubility for ML395 in PBS was determined to be >94.5±0.8 μM (>42.3 μg/mL), which is 263-fold higher than the cellular IC50 for PLD2.
GSH Conjugates. No glutathione conjugates detected.
Stability. Stability was determined for ML395 at 23 °C in PBS (no antioxidants or other protectorants and DMSO concentration below 0.1%). After 48 hours, ∼100% of the initial concentration of ML395 remained in solution. Thus, ML395 is very stable in vitro in assay buffer as well as in vivo (vide infra). In addition, ML395 was incubated in rat plasma for 3 hours at 37 °C, and was found to be stable – no degradation.
| Percent Remaining (%) | ||||||
|---|---|---|---|---|---|---|
| Compound | 0 Min | 15 Min | 30 Min | 90 min. | 24 Hour | 48 Hour |
| ML395, CID 73099363 | 100 | 99.2 | 99.8 | 100 | 101 | 102 |
Compounds added to the SMR collection (MLS#s): MLS005886367 (ML395, CID 73099418 –free base, 21.9 mg); MLS005886368 (CID 73099417, 5.7 mg); MLS005886369 (CID 73099472, 10 mg); MLS005886370 (CID 73099378, 6.9 mg); MLS005886371 (CID 73099437, 8.3 mg); MLS005886372 (CID 73099333, 9.0 mg).
2.3. Probe Preparation
General Experimental
All reagents were purchased from commercial suppliers and were purified as needed according to the procedures of Armarego and Chai. Analytical thin-layer chromatography (TLC) was performed on 250 μm silica plates from Sorbent Technologies. Visualization was accomplished via UV light, and/or the use of ninhydrin, iodine, and potassium permanganate solutions followed by application of heat. Chromatography was performed using Silica Gel 60 (230-400 mesh) from Sorbent Technologies or Silica RediSep Rf flash columns on a CombiFlash Rf automated flash chromatography system. All 1H and 13C NMR spectra were recorded on a Bruker AV-400 (400 MHz) instrument. Chemical shifts are reported in ppm relative to residual solvent peaks as an internal standard set to δ 7.26 and δ 77.16 (CDCl3) or δ 3.31 and δ 49.00 (MeOD). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, sx = sextet, sp = septet, br = broad, dd = doublet of doublets, dq = doublet of quartets, td = triplet of doublets, pd = pentet of doublets, m = multiplet), coupling constant (Hz), integration. Low resolution mass spectra (LCMS) were recorded on an Agilent 1200 LCMS with electrospray ionization. High resolution mass spectra (HRMS) were recorded on a Waters QToF-API-US plus Acuity system with ES as the ion source. Analytical high performance liquid chromatography (HPLC) was performed on an Agilent 1200 analytical LCMS with UV detection at 214 nm and 254 nm along with ELSD detection.
N-(2-(4-oxo-1-(pyridine-3-ylmethyl)-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)2-naphthamide:, ML395: To an oven dried round-bottomed flask equipped with a magnetic stir bar under argon was added triazaspirone 28 starting material (1.00 g, 4.08 mmol, 1.00 eq.), diisopropyl ethyl amine (2.63 g, 3.55 mL, 20.38 mmol, 5.00 eq.), 4-dimethylaminopyridine (0.25 g, 2.04 mmol, 0.50 eq.), and those contents were dissolved in THF (41 mL). Di-tert-butyl dicarbonate (0.98 g, 4.48 mmol, 1.10 eq) was added, as a liquid, to the mixture at room temperature, and the reaction was stirred for 1.5 hours at room temperature. The solvent was then removed under reduced pressure, and the residual material was dissolved in diethyl ether (150 mL). That organic layer was washed with saturated brine (2 × 100 mL), dried with magnesium sulfate, filtered, and concentrated under reduced pressure to provide 1.487 g of crude Boc'd material that was used in the next reaction without further purification. Crude Boc'd 28 (1.49 g), in a round-bottomed flask equipped with a magnetic stir bar, from the previous reaction was dissolved in methanol (82 mL), and 10% palladium on carbon (0.22 g, 0.20 mmol, 0.05 eq) was added to the solution under argon. The reaction was purged with hydrogen gas and was stirred under a hydrogen atmosphere for 15 hours. The reaction was filtered through a celite pad to remove the palladium on carbon, and the filtrate was concentrated under reduced pressure to provide 1.10 g of crude secondary amine, which was used in the next reaction without further purification. Crude secondary amine (1.10 g), in a flame dried round-bottomed flask equipped with a magnetic stir bar, from the previous reaction was dissolved in DMF (8.2 mL). Potassium carbonate (2.82 g, 20.39 mmol, 5.00 eq) followed by tert-butyl (2-bromoethyl) carbamate (1.325 g, 4.91 mmol, 1.45 eq) were added to the reaction mixture at room temperature and it was stirred for 22 hours. The reaction was then partitioned between with ether (50 mL) and water (50 mL). The reaction mass was extracted with ether (3 × 50 mL), the combined organic was dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The crude material was purified using flash column chromatography (0-20% methanol (w/10% NH4OH) in ethyl acetate, stain with KMnO4) to yield 1.01 g (62% over 3 steps) of the desired product 29. 1H NMR (400.1 MHz, MeOD) δ (ppm): 4.58 (s, 2H); 3.20 (t, J = 6.7 Hz, 2H); 2.83 (dt, J1 = 11.8 Hz, J2 = 3.6 Hz, 2H); 2.50 (t, J = 6.8 Hz, 2H); 2.39 (td, J1 = 12.0 Hz, J2 = 2.2 Hz, 2H); 1.91 (td, J1 = 13.7 Hz, J2 = 4.2 Hz, 2H); 1.65 (d, J = 13.1 Hz, 2H); 1.52 (s, 9H); 1.44 (s, 9H). 13C NMR (100.6 MHz, MeOD) δ (ppm): 178.67, 158.39, 150.64, 84.45, 80.06, 62.41, 61.57, 58.73, 49.92, 38.44, 31.66, 28.75, 28.19. HRMS (TOF, ES+) C19H35N4O5 [M+H]+ calc. mass 399.2607, found 399.2605.
To a solution of starting material 29 (0.94 g, 2.36 mmol, 1.00 eq) in dichloromethane (24 mL) was added 4 M HCl in dioxane (17.7 mL, 70.84 mmol, 30.00 eq), at room temperature all at once. The reaction was stirred for 18 hours at room temperature at which point the reaction was concentrated under reduced pressure to afford the tris-HCl salt 30 (0.726 g, quantitative yield). The product 30 was used in the next reaction without further purification. The crude starting material 30 (0.726 g, 2.36 mmol, 1.00 eq) from the previous reaction, phthalic anhydride (0.524 g, 3.54 mmol, 1.50 eq), and diisopropyl ethyl amine (1.52 g, 2.06 mL, 11.80 mmol, 5.00 eq) were added to a microwave vial and dissolved in DMF (11.8 mL). The reaction was heated in a microwave reactor at 120 °C for 20 minutes. The crude mixture in DMF was injected onto a reverse phase HPLC (5-20% acetonitrile in water (0.1% trifluoroacetic acid)) for purification monitoring product elution at 221 nm to provide the bis trifluoroacetic acid salt 31 (956 mg, 73%). 1H NMR (400.1 MHz, MeOD) δ (ppm): 7.93-7.88 (m, 2H); 7.87-7.81 (m, 2H); 4.57 (s, 2H); 4.13 (t, J = 5.89 Hz, 2H); 3.90-3.41 (m, 6H); 2.50-2.04 (m, 4H). 13C NMR (100.6 MHz, MeOD) δ (ppm): 168.35, 160.26, 134.20, 131.88, 123.03, 57.17, 54.72, 48.38, 31.97, 27.70. HRMS (TOF, ES+) C17H21N4O3 [M+H]+ calc. mass 329.1614, found 326.1617.
A mixture of starting material 31 (0.37 g, 0.66 mmol, 1.00 eq) and 3-pyridine carboxaldehyde (0.36 g, 3.32 mmol, 5.00 eq) were stirred in glacial acetic acid (3.3 mL) for 20 minutes. Sodium triacetoxy borohydride (0.703 g, 3.32 mmol, 5.00 eq) was added to the mixture, and it was left to stir at room temperature for an additional 30 minutes. The acetic acid was then removed via vacuum distillation and the dry reaction mass was resuspended in methanol (10 mL) and concentrated under reduced pressure to remove boron byproducts as trimethyl borate. The crude material was then purified via flash column chromatography (10-20% methanol in ethyl acetate) to afford the product 32 (0.254 g, 91%). 1H NMR (400.1 MHz, MeOD) δ (ppm): 8.56-8.37 (m, 2H); 7.89-7.76 (m, 5H); 7.40 (dd, J1 = 7.8 Hz, J2 = 4.9 Hz, 1H); 3.98-3.91 (m, 4H); 3.79 (s, 2H); 3.30-3.23 (m, 2H); 3.17 (td, J1 = 11.7 Hz, J2 = 2.9 Hz, 2H); 3.02 (t, J = 6.2 Hz, 2H); 2.10-2.00 (m, 2H); 1.95-1.87 (m, 2H). 13C NMR (100.6 MHz, MeOD) δ (ppm): 179.37, 169.69, 150.06, 149.05, 138.41, 136.06, 135.27, 133.48, 125.23, 124.15, 62.24, 60.14, 56.20, 50.30, 48.72, 35.09, 28.90. HRMS (TOF, ES+) C23H26N5O3 [M+H]+ calc. mass 420.2036, found 420.2034.
To a mixture of starting material 32 (0.471 g, 1.12 mmol, 1.00 eq) in acetonitrile (9.4 mL) was added hydrazine (0.360 g, 11.23 mmol, 10.00 eq). The reaction was stirred for 15 hours before the acetonitrile was removed under reduced pressure. The solid material was redissolved in methanol and was passed through a cation exchange cartridge. The product 33 was eluted from the column with 2 M NH4OH in methanol, and the filtrate was concentrated under reduced pressure to afford the product 33 as a crude solid (0.322 g, 99%) that was used without further purification. Crude starting material 33 (0.12 g) and diisopropyl ethyl amine (0.266 g, 0.364 mL, 2.06 mmol, 5.00 eq), in a flame dried round-bottomed flask equipped with a magnetic stir bar, were dissolved in DMF (4.1 mL). To that flask was added naphthoyl chloride (0.10 g, 0.49 mmol, 1.20 eq), and the reaction was stirred for 30 minutes. The crude mixture was injected onto a reverse phase HPLC (5-80% acetonitrile in water (0.1% NH4OH)) for purification monitoring product elution at 215 nm to provide the solid product ML395 (34) (137 mg, 75%). 1H NMR (400.1 MHz, MeOD) δ (ppm): 8.53-8.36 (m, 3H); 8.01-7.72 (m, 6H); 7.56-7.47 (m, 2H); 7.34 (dd, J1 = 7.7 Hz, J2 = 4.9 Hz, 1H); 3.88 (s, 2H); 3.72 (s, 2H); 3.61 (t, J = 6.8 Hz, 2H); 3.35 (s, 1H); 2.92-2.79 (m, 4H); 2.69 (t, J = 6.8 Hz, 2H); 1.89 (td, J1 = 13.5 Hz, J2 = 5.0 Hz, 2H); 1.82-1.73 (m, 2H). 13C NMR (100.6 MHz, MeOD) δ (ppm): 179.96, 169.99, 150.08, 149.00, 138.37, 136.23, 136.14, 133.96, 132.81, 129.97, 129.29, 128.79, 128.77, 128.74, 127.81, 125.19, 124.87, 62.19, 60.88, 58.03, 50.48, 48.85, 38.34, 29.66. HRMS (TOF, ES+) C26H30N5O2 [M+H]+ calc. mass 444.2400, found 444.2402.
3. Results
This is a Medicinal Chemistry Fast-Track entry into the MLPCN network, and the third probe submitted. Utilizing the halopemide scaffold 22 as a basis for ligand design, multi-dimensional libraries (∼800 compounds to date) surveying three different regions of the scaffold led to the discovery of A diversity-oriented synthesis (DOS) approach led to the identification of a number of highly selective PLD1 inhibitors such as VU0359595 (23). From this, a key triazaspirone moiety was discovered that engendered preferential PLD2 inhibition, as shown in VU0285655 (24). A 4 × 6 member library then led to the discovery of a highly selective PLD2 inhibitor VU0364739 (25), displaying an unprecedented 75-fold selectivity. However, 25 still affords potent inhibition of PLD1 (IC50 = 1,500 nM), and at standard 10 μM in vitro studies and in vivo plasma exposures, both PLD1 and PLD2 will be inhibited. Thus, there was significant room for improvement. Moreover, only 4 amides were surveyed in the context of the more optimal 1-(3-fluorophenyl)-1,3,8-triazspiro[4.5]decan-4-one 26, and future work would evaluate additional amides. Furthermore, the benzimidazolone moiety engendered more ancillary pharmacology at biogeneic amines than the triazaspirone core, and thus represents a more attractive starting point for further optimization of both PLD1 and PLD2 inhibitors. Further chemical optimization, through the iterative parallel synthesis of another 28 amides, led to the discovery of ML298, a >53-fold PLD2 selective inhibitor (PLD2 IC50 = 355 nM), with no activity at PLD1 (IC50 >20,000 nM) and modest DMPK properties and in vivo exposure; moreover, we also developed ML299, a potent dual PLD1/2 inhibitor. Despite the advance, these compounds suffered from solubility issues, low free fraction, modest PK and toxicity at high concentrations. Further optimization of ML298 led to the development of a new chemotype, represented by ML395, a potent and selective PLD2 inhibitor (PLD2 IC50 = 360 nM, PLD1 IC50 >30,000 nM, >80-fold selective) with improved free fraction, PK, solubility and no observed toxicity. In addition, the key structural modification eliminated all biogenic amine activity. The cell-based, functional PLD1 and PLD2 assays were the work-horse primary assays, with the biochemical PLD1 and PLD2 assays as secondary to confirm a direct interaction with the PLD proteins. ML395 enabled our labs to study the role of selective PLD2 inhibition in cellular models of influenza infection.
3.1. Dose Response Curves for Probe
In both cellular assays and biochemical inhibition assays with purified PLD1 and PLD2, ML395 preferentially inhibited PLD2 over PLD1 with a cellular IC50 of 360 nM and PLD1 with an IC50 of >30,000 nM, and in the purified biochemical assay with a PLD2 IC50 of 8,7000 nM and PLD1 with an IC50 of >30,000 nM (Figure 4).2 In both assays, ML395 was more potent at inhibiting PLD2 (>80-fold PLD2 selective in cells) than PLD1. ML395 is the first isoform selective PLD2 inhibitor that does not inhibit PLD1 at concentrations routinely used for in vitro studies, and will not inhibit PLD1 when dosed in vivo. Importantly, the agreement between the two assays in terms of potency and isoform selectivity indicates that ML395 is acting directly on the PLD proteins (Figure 4B).

The primary screening assays are cell-based assays, indicating that ML395 can gain access to its molecular target when applied to cells. The compound did not exhibit acute toxicity in cell based assays at concentrations up to 50 μM, and cyctotoxicity assays aimed at this parameter indicated ML395 had no cyctotoxicity in non-transformed HEK293 cells as well as A549 cells. ML395 has also been tested in multiple cancer cell lines.

Figure 5Other PLD2 inhibitors such as 25 induce significant cytotoxicity above 10 μM concentrations in A549 epithelial cells, whereas ML395 shows no cytotoxicity up to 50 μM
3.2. Profiling Assays
To more fully characterize this potent and highly PLD2 selective inhibitor, ML395 was tested using Eurofins Panlabs's (formerly MDS Pharma's) Lead Profiling Screen (binding assay panel of 68 GPCRs, ion channels and transporters screened at 10 μM).45 Included in the Eurofins Panlabs screening panel are a number of ion channels (Calcium Channel, L-Type and N-Type; Potassium channel [KATP]; Potassium channel [hERG]) and class A GPCRs (D1-5, H1-3, etc…). ML395 was found to potently bind in only 2 of the 68 assays conducted (inhibition of radio ligand binding > 70% at 10 μM; Table 3). ML395, while much cleaner than halopemide (activity at 41 of 68 targets) and improved relative to other isoform selective PLD inhibitors, possessing no binding affinity at CNS biogenic amine targets. ML395 displaced the radioligands for 5-HT2B and sodium channel type 2: however, in follow-up functional assays, ML395 possessed no functional activity. We also performed a kinome scan, and ML395 was inactive against all kinases in a broad kinase panel. Table 4 highlights calculated properties for ML395, which compares favorably with the MDDR.
Table 3
Eurofins Panlabs Profiling of ML395.
Table 4
Calculated Property Comparison with MDDR Compounds.
In order to aid the wider community in the use ML395, we further profiled ML395 in a battery of pharmacokinetic assays (Table 5) including an assessment of intrinsic clearance (CLINT) in hepatic microsomes, allowing for the prediction of pertinent rat and human PK parameters (CL and t1/2). ML298 is predicted to have high clearance in both rat (CLINT = 82.1mL/min/kg, CLHEP = 64.3 mL/min/kg) and human (CLINT = 43.1 mL/min/kg, CLHEP = 17.2 mL/min/kg), and possesses exceptional free fraction (8.8% free in human and 26.3% free in rat (equilibrium dialysis plasma protein binding studies)). To ensure that the free fraction was real and not due to plasma instability, ML395 was incubated in rat plasma for 3 hours at 37 °C, and ML395 was stable – 100% parent remained. Thus, ML395 represents a 10-fold improvement in free fraction in rat over ML298, and should be a better in vivo tool. Moreover, ML395 has a variable CYP P450 profile, displaying inhibition of two of the four major CYPs (3A4 (3.9 μM), 2D6 (16.2 μM), 2C9 (>30 μM) and 1A2 (>30 μM)).
Table 5
In vitro DMPK Profile of ML395.
When dosed IP in a plasma/brain study, ML395 afforded excellent brain exposure (BrainAUC/PlasmaAUC of 1.48), as opposed to ML298 with a B:P ratio of 0.005; thus, we had developed a highly brain penetrant PLD2 inhibitor – the first such tool to dissect PLD2 function in the CNS. Therefore, ML395 can be used as both an in vitro and in vivo probe for CNS indications such as PD and AD.
4. Discussion
4.1. Comparison to existing art and how the new probe is an improvement
ML395 is a potent, selective and direct inhibitor of PLD2 in cells (PLD2, IC50 = 360 nM and PLD1, IC50 = >30,000 nM) as well as in a biochemical assay with purified PLD2 (PLD2, IC50 = 8,700 nM) and PLD1 (PLD1, IC50 >20,000 nM). Moreover, ML395 is the most selective (>80-fold over PLD1) PLD2 inhibitor ever described that does not inhibit PLD1 at standard in vitro doses and in vivo exposure levels. ML395 possesses an acceptable in vitro and in vivo DMPK profile for preliminary proof of concept studies for both cancer and virology, and unlike earlier probes, displays no toxicity in cells up to 30 μM! ML395 is stable in rat plasma, has a true free fraction of over 25% and is highly CNS penetrant (B:P = 1.48). ML395 is a major improvement over the classical tool to study PLD function, n-butanol. Again, n-butanol is not a PLD inhibitor, rather n-butanol (and some other primary alcohols) block PLD-catalyzed phosphatidic acid production by competing with water as a nucleophile thereby causing the formation of phosphatidylbutanol. In addition, there are concerns that n-butanol may not fully block PA production and n-butanol may also be promiscuous in cell-based assays affecting multiple pathways in addition to transphosphatidylation. Thus, conclusions reached in the literature from studies employing n-butanol alone should be viewed with caution, and ML395 provides the means to address these issues, as well as providing isoform selective PLD2 inhibition. In addition, ML395 represents and improvement over halopemide (22), as 22 has potent activity at D2 and over 40 other biogenic amine receptors. Finally, ML395 is free from IP constraints, which will allow the MLPCN to freely provide this probe to the biomedical research community, and over two grams of this probe has already been shipped to St. Jude's for in vivo infection models in mice.
5. References
- 1.
- Selvy PE, Lavieri R, Lindsley CW, Brown HA. ‘Phospholipase D: enzymology, signaling and chemical modulation. Chem. Rev. 2011;111:6064–6119. [PMC free article: PMC3233269] [PubMed: 21936578]
- 2.
- Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga CL, Lindsley CW, Brown HA. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat Chem Biol. 2009;5:108–117. [PMC free article: PMC3798018] [PubMed: 19136975]
- 3.
- Brown HA, Henage LG, Preininger AM, Xiang Y, Exton JH. Biochemical analysis of phospholipase D. Methods Enzymol. 2007;434:49–87. [PubMed: 17954242]
- 4.
- Foster DA. Phosphatidic acid signaling to mTOR: Signals for the survival of human cancer cells. Biochim Biophys Acta. 2009;1791:949–955. [PMC free article: PMC2759177] [PubMed: 19264150]
- 5.
- Noh DY. Overexpression of phospholipase D1 in human breast cancer tissues. Cancer Lett. 2000;161:207–214. [PubMed: 11090971]
- 6.
- Zhao Y, Ehara H, Akao Y, Shamoto M, Nakagawa Y, Banno Y, Deguchi T, Ohishi N, Yagi K, Nozawa Y. Increased activity and intranuclear expression of phospholipase D2 in human renal cancer. Biochem Biophys Res Commun. 2000;278:140–143. [PubMed: 11185526]
- 7.
- Yamada Y. Association of a polymorphism of the phospholipase D2 gene with the prevalence of colorectal cancer. J. Mol. Med. 2003;81:126–131. [PubMed: 12601529]
- 8.
- Oliveira TG, Chan RB, Tian H, Laredo M, Shui G, Staniszewski A, Zhang H, Wang L, Kim TW, Duff KE, Wenk MR, Arancio O, Di Paolo G. Phospholipase D2 Ablation Ameliorates Alzheimer's Disease-Linked Synaptic Dysfunction and Cognitive Deficits. J. Neurosci. 2010;30:16419–16428. [PMC free article: PMC3004537] [PubMed: 21147981]
- 9.
- Elvers M, Stegner D, Hagedorn I, Kleinschnitz C, Braun A, Kuijpers ME, Boesl M, Chen Q, Heemskerk JW, Stoll G, Frohman MA, Nieswandt B. Impaired αIIbβ3 Integrin Activation and Shear-Dependent Thrombus Formation in Mice Lacking Phospholipase D1. Sci. Signal. 2010;3:1–10. [PMC free article: PMC3701458] [PubMed: 20051593]
- 10.
- Karlas A, Machuy N, Shin Y, Pleissner K-P, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, Mäurer AP, Müller E, Wolff T, Rudel T, Meyer TF. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–825. [PubMed: 20081832]
- 11.
- Chu M, Patel MG, Pai J-K, Das PR, Puar MS. Sch 53823 and Sch 53825, novel fungal metabolites with phospholipase D inhibitory activity. Bioorganic & Medicinal Chemistry Letters. 1996;6:579–584.
- 12.
- Hegde VR, Silver J, Patel MG, Bryant R, Pai J, Das PR, Puar MS, Cox PA. Phospholipase D inhibitors from a Myrsine species. J Nat Prod. 1995;58:1492–7. [PubMed: 8676128]
- 13.
- Clark AM, El-Feraly FS, Li WS. Antimicrobial activity of phenolic constituents of Magnolia grandiflora L. J Pharm Sci. 1981;70:951–2. [PubMed: 7310672]
- 14.
- Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, Der CJ, Battle T, Frank DA, Ye K, Murad E, Dubiel W, Soff G, Arbiser JL. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278:35501–7. [PubMed: 12816951]
- 15.
- Shigemura K, Arbiser JL, Sun SY, Zayzafoon M, Johnstone PA, Fujisawa M, Gotoh A, Weksler B, Zhau HE, Chung LW. Honokiol, a natural plant product, inhibits the bone metastatic growth of human prostate cancer cells. Cancer. 2007;109:1279–89. [PubMed: 17326044]
- 16.
- Garcia A, Zheng Y, Zhao C, Toschi A, Fan J, Shraibman N, Brown HA, Bar-Sagi D, Foster DA, Arbiser JL. Honokiol suppresses survival signals mediated by Ras-dependent phospholipase D activity in human cancer cells. Clin Cancer Res. 2008;14:4267–74. [PMC free article: PMC2759181] [PubMed: 18594009]
- 17.
- Tou JS, Urbizo C. Diethylstilbestrol inhibits phospholipase D activity and degranulation by stimulated human neutrophils. Steroids. 2008;73:216–21. [PubMed: 18036628]
- 18.
- Kitzen JJ, de Jonge MJ, Lamers CH, Eskens FA, van der Biessen D, van Doorn L, Ter Steeg J, Brandely M, Puozzo C, Verweij J. Phase I dose-escalation study of F60008, a novel apoptosis inducer, in patients with advanced solid tumours. Eur J Cancer. 2009;45:1764–72. [PubMed: 19251409]
- 19.
- Kang DW, Lee JY, Oh DH, Park SY, Woo TM, Kim MK, Park MH, Jang YH, Min do S. Triptolide-induced suppression of phospholipase D expression inhibits proliferation of MDA-MB-231 breast cancer cells. Exp Mol Med. 2009;41:678–85. [PMC free article: PMC2753661] [PubMed: 19478552]
- 20.
- Westerheide SD, Kawahara TL, Orton K, Morimoto RI. Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J Biol Chem. 2006;281:9616–22. [PubMed: 16469748]
- 21.
- Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, Goodrich JA, Liu JO. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol. 2011 [PMC free article: PMC3622543] [PubMed: 21278739]
- 22.
- Leiros I, Secundo F, Zambonelli C, Servi S, Hough E. The first crystal structure of a phospholipase D. Structure. 2000;8:655–67. [PubMed: 10873862]
- 23.
- Davies DR, Interthal H, Champoux JJ, Hol WG. Insights into substrate binding and catalytic mechanism of human tyrosyl-DNA phosphodiesterase (Tdp1) from vanadate and tungstate-inhibited structures. J Mol Biol. 2002;324:917–32. [PubMed: 12470949]
- 24.
- Davies DR, Interthal H, Champoux JJ, Hol WG. Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide. Chem Biol. 2003;10:139–47. [PubMed: 12618186]
- 25.
- McDonald LA, Barbieri LR, Bernan VS, Janso J, Lassota P, Carter GT. 07H239-A, a new cytotoxic eremophilane sesquiterpene from the marine-derived xylariaceous fungus LL-07H239. Journal of Natural Products. 2004;67:1565–1567. [PubMed: 15387660]
- 26.
- Puar MS, Barrabee E, Hallade M, Patel M. Sch 420789: A novel fungal metabolite with phospholipase D inhibitory activity. J Antibiot. 2000;53:837–838. [PubMed: 11079806]
- 27.
- Diwu Z, Zimmermann J, Meyer T, Lown JW. Design, synthesis and investigation of mechanisms of action of novel protein kinase C inhibitors: perylenequinonoid pigments. Biochem Pharmacol. 1994;47:373–85. [PubMed: 7508231]
- 28.
- Sciorra VA, Hammond SM, Morris AJ. Potent direct inhibition of mammalian phospholipase D isoenzymes by calphostin-c. Biochemistry. 2001;40:2640–2646. [PubMed: 11327888]
- 29.
- Levy BD, Hickey L, Morris AJ, Larvie M, Keledjian R, Petasis NA, Bannenberg G, Serhan CN. Novel polyisoprenyl phosphates block phospholipase D and human neutrophil activation in vitro and murine peritoneal inflammation in vivo. British Journal of Pharmacology. 2005;146:344–351. [PMC free article: PMC1440714] [PubMed: 16041402]
- 30.
- Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci. 2008;65:1631–52. [PMC free article: PMC4686230] [PubMed: 18324353]
- 31.
- Yamamoto H, Hanada K, Kawasaki K, Nishijima M. Inhibitory effect on curcumin on mammalian phospholipase D activity. FEBS Lett. 1997;417:196–8. [PubMed: 9395294]
- 32.
- Jaiyesimi IA, Buzdar AU, Decker DA, Hortobagyi GN. Use of tamoxifen for breast cancer: twenty-eight years later. J Clin Oncol. 1995;13:513–29. [PubMed: 7844613]
- 33.
- Perry RR, Kang Y, Greaves B. Effects of tamoxifen on growth and apoptosis of estrogen-dependent and -independent human breast cancer cells. Ann Surg Oncol. 1995;2:238–45. [PubMed: 7641021]
- 34.
- Noh DY. Overexpression of phospholipase D1 in human breast cancer tissues. Cancer Lett. 2000;161:207–214. [PubMed: 11090971]
- 35.
- Eisen SF, Brown HA. Selective estrogen receptor (ER) modulators differentially regulate phospholipase D catalytic activity in ER-negative breast cancer cells. Mol Pharmacol. 2002;62:911–20. [PubMed: 12237338]
- 36.
- Borgna JL, Rochefort H. Hydroxylated metabolites of tamoxifen are formed in vivo and bound to estrogen receptor in target tissues. J Biol Chem. 1981;256:859–68. [PubMed: 7451477]
- 37.
- Desta Z, Ward BA, Soukhova NV, Flockhart DA. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6. J Pharmacol Exp Ther. 2004;310:1062–75. [PubMed: 15159443]
- 38.
- Jordan VC, Collins MM, Rowsby L, Prestwich G. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J Endocrinol. 1977;75:305–16. [PubMed: 591813]
- 39.
- Coezy E, Borgna JL, Rochefort H. Tamoxifen and metabolites in MCF7 cells: correlation between binding to estrogen receptor and inhibition of cell growth. Cancer Res. 1982;42:317–23. [PubMed: 7053859]
- 40.
- Kiss Z, Anderson WH. Inhibition of phorbol ester-stimulated phospholipase D activity by chronic tamoxifen treatment in breast cancer cells. FEBS Lett. 1997;400:145–8. [PubMed: 9001386]
- 41.
- Monovich L, Mugrage B, Quadros E, Toscano K, Tommasi R, LaVoie S, Liu E, Du ZM, LaSala D, Boyar W, Steed P. Optimization of halopemide for phospholipase D2 inhibition. Bioorganic & Medicinal Chemistry Letters. 2007;17:2310–2311. [PubMed: 17317170]
- 42.
- De Cuyper H, van Praag HM, Verstraeten D. The Clinical Significance of Halopemide, a Dopamine-Blocker Related to the Butyrophenones. Neuropsychobiology. 1984;12:211–223. [PubMed: 6398861]
- 43.
- Lavieri R, Lewis JA, Scott SA, Selvy PE, Buck J, Armstrong MD, Brown HA, Lindsley CW. ‘Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part II: PLD2 selectivity by virtue of a triazaspirone privileged structure’ Bioorg. Med.Chem. Lett. 2009;19:2240–2244. [PMC free article: PMC3800051] [PubMed: 19299128]
- 44.
- Lavieri R, Scott SA, Selvy PE, Brown HA, Lindsley CW. ‘Design, Synthesis and Biological Evaluation of Halogenated N-(2-(4-Oxo-1-phenyl-1,3,8-triazasprio[4.5]decan-8-yl)ethylbenzamides: Discovery of an Isoform Selective Small-Molecule Phospholipase D2 (PLD2) Inhibitor’ J. Med. Chem. 2010;53:6706–6719. [PMC free article: PMC3179181] [PubMed: 20735042]
- 45.
- For information on the Eurofins Panlabs lead profiling screen see: www
.EurofinsPanlabs.com
- PMCPubMed Central citations
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Review Development of a Selective, Allosteric PLD2 Inhibitor.[Probe Reports from the NIH Mol...]Review Development of a Selective, Allosteric PLD2 Inhibitor.Scott SA, O’Reilly MC, Daniels JS, Morrison R, Ptak R, Dawson ES, Tower N, Engers JL, Engers DW, Oguin T, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Development of a Selective, Allosteric PLD1/2 Inhibitor in a Novel Scaffold.[Probe Reports from the NIH Mol...]Review Development of a Selective, Allosteric PLD1/2 Inhibitor in a Novel Scaffold.Scott SA, O’Reilly MC, Daniels JS, Morrison R, Ptak R, Dawson ES, Tower N, Engers JL, Engers DW, Oguin T, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Discovery of a highly selective PLD2 inhibitor (ML395): a new probe with improved physiochemical properties and broad-spectrum antiviral activity against influenza strains.[ChemMedChem. 2014]Discovery of a highly selective PLD2 inhibitor (ML395): a new probe with improved physiochemical properties and broad-spectrum antiviral activity against influenza strains.O'Reilly MC, Oguin TH 3rd, Scott SA, Thomas PG, Locuson CW, Morrison RD, Daniels JS, Brown HA, Lindsley CW. ChemMedChem. 2014 Dec; 9(12):2633-7. Epub 2014 Sep 10.
- Review Development of a Second Generation mGlu(3) NAM Probe.[Probe Reports from the NIH Mol...]Review Development of a Second Generation mGlu(3) NAM Probe.Wenthur C, Daniels JS, Morrison R, Engers JL, Niswender CM, Conn PJ, Lindsley CW. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Development of the First Selective mGlu(3) NAM from an mGlu(5) PAM Hit.[Probe Reports from the NIH Mol...]Review Development of the First Selective mGlu(3) NAM from an mGlu(5) PAM Hit.Sheffler DJ, Wenthur CJ, Brunner JA, Daniels JS, Morrison RD, Blobaum AL, Dawson ES, Engers JL, Niswender CM, Conn PJ, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- A Next generation PLD2 inhibitor with improved physiochemical properties and DMP...A Next generation PLD2 inhibitor with improved physiochemical properties and DMPK profile for translational in vivo - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...