Summary
The purpose of this overview is to increase the awareness of clinicians regarding congenital diaphragmatic hernia and its genetic causes and management. The following are the goals of this overview.
Goal 1.
Describe the clinical characteristics of congenital diaphragmatic hernia.
Goal 2.
Review the genetic causes of congenital diaphragmatic hernia.
Goal 3.
Provide an evaluation strategy to identify the genetic cause of congenital diaphragmatic hernia in a proband (when possible).
Goal 4.
Review management of congenital diaphragmatic hernia.
Goal 5.
Inform genetic counseling of family members of an individual with congenital diaphragmatic hernia.
1. Clinical Characteristics of Congenital Diaphragmatic Hernia
Clinical Description
Congenital diaphragmatic hernia (CDH) refers to a developmental defect of the formation of the diaphragm that, in most individuals, is evident at birth. CDH is characterized by: (1) incomplete formation/muscularization of the diaphragm resulting in absence or deficiency of the diaphragm, or (2) eventration resulting in elevation of a portion of the diaphragm that is thinned as a result of incomplete muscularization. The prevalence of CDH is estimated at 3-3.6/10,000 live births [Wang et al 2011, Wright et al 2011, Stoll et al 2015, Burgos & Frenckner 2017, Shanmugam et al 2017].
Infants with CDH often present in the neonatal period with severe respiratory distress.
Presentation after infancy, occurring in 5%-10% of affected individuals, includes respiratory distress, such as from pleural effusion due to entrapment of the bowel in the chest, or gastrointestinal distress, such as abdominal pain from chronic or intermittent intestinal obstruction. About 1% of individuals are completely asymptomatic and the defect is discovered incidentally on imaging studies [Bagłaj & Dorobisz 2005].
At least 10% of individuals reherniate following initial surgical repair; the risk is considerably greater among those whose hernia repair required a prosthetic patch.
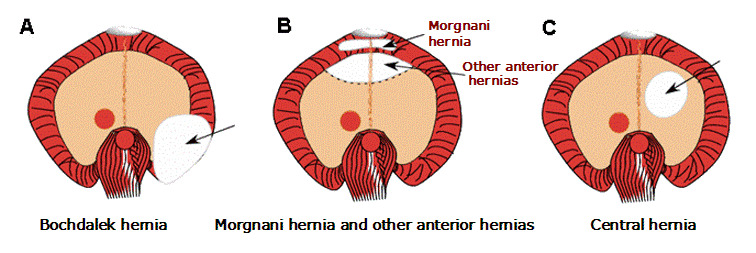
Although CDH is classified into several types, distinction among hernias can be problematic. An anatomic depiction of the normal diaphragm is presented in Figure 1 and anatomic descriptions of diaphragmatic defects are presented in Figure 2.

Figure 1
A. Normal diaphragm (for reference) B. View of normal diaphragm from below

Figure 2.
View of diaphragmatic defects from below A. Bochdalek hernia
Posterolateral (Bochdalek) hernia of the diaphragm is often accompanied by herniation of the stomach, intestines, liver, and/or spleen into the chest cavity. An extremely large defect, or apparent absence of the hemidiaphragm, is called agenesis of the diaphragm; this defect probably represents the severe end of the Bochdalek hernia spectrum.
Posterolateral hernias comprise approximately 80%-90% of all CDH and appear to fall into two types:
- A diaphragmatic defect with an intact rim of posterior and lateral musculature
About 85% of Bochdalek hernias occur on the left side, about 10% on the right, and approximately 5% are bilateral.
Non-posterolateral (non-Bochdalek) hernia. Anterior defects of the diaphragm can occur in the midline, on the left side, or the right side. Several distinct subtypes are described, with considerable overlap in the anatomic location.
- Morgagni (Morgagni-Larrey) hernia is an anterior retrosternal or parasternal hernia that can result in the herniation of liver or intestines into the chest cavity (see Figure 2B). Morgagni hernias comprise approximately 2% of all CDH, are generally accompanied by a hernia sac, and often do not cause symptoms in the newborn period.
- Other anterior hernias associated with pentalogy of Cantrell. These rare and severe types of hernia are found in individuals with pentalogy of Cantrell (which also includes defects in the supraumbilical midline abdominal wall, lower sternum, diaphragmatic pericardium, and heart).
- Central hernia. This hernia is a rare diaphragm defect involving the central tendinous (i.e., amuscular) portion of the diaphragm. The entire rim of diaphragmatic musculature is present (see Figure 2C).
Diaphragmatic eventration is incomplete muscularization of the diaphragm resulting in a thin membranous sheet of tissue. It is difficult to estimate the frequency of eventration because it may coexist with and/or be misdiagnosed as a Bochdalek hernia. Severe diaphragmatic eventration is associated with pulmonary hypoplasia and respiratory distress during infancy. Milder degrees of diaphragmatic eventration can present later in life with respiratory symptoms such as cough and pneumonias, or without symptoms, so that the diagnosis is made incidentally on chest radiograph. Increasingly, it is observed that eventration of the diaphragm and CDH can occur in the same individual and in different individuals with pathogenic variants in the same gene, suggesting that in some instances they share a common etiology.
Sac hernia. All hernia types can present with a sac (e.g., membranous sheet of tissue) covering the herniated abdominal contents. Animal models suggest that abnormal connective tissue, resulting in a localized amuscular region, rather than a primary defect in myogenic cells, is the embryogenetic basis of the hernia sac [Merrell et al 2015]. The presence of a sac is often reported to be associated with a better prognosis. However, because a thin and redundant membranous diaphragm resulting from an eventration defect may represent a "sac," it is probable that eventration and "sac type" CDH diagnoses are often interchanged. For this reason, it is inaccurate to assume that the prognosis is better when a sac is visualized.
CDH can occur as either an isolated or complex anomaly:
- Isolated. Observed in 50%-60% of probands; not associated with other major congenital anomalies
- Complex. Observed in 40%-50% of probands and associated with other congenital anomalies:
- Syndromic. Some individuals with complex CDH have a readily identifiable genetic syndrome or chromosome anomaly (see Tables 1, 2, and 3). Individuals with syndromic CDH are more likely to be diagnosed prenatally, have an affected first-degree relative (10%), and have a worse prognosis. More than 50% of individuals with syndromic CDH are born preterm and have low birth weight [Shanmugam et al 2017].
- Complex CDH not attributed to a recognized syndrome. The remaining probands with complex CDH present with associated additional malformations (e.g., craniofacial, ocular, cardiovascular, central nervous system, limb, genitourinary) but do not have a recognizable syndromic phenotype.
Establishing the Diagnosis of CDH
Congenital diaphragmatic hernias are increasingly diagnosed prenatally; however, they are occasionally diagnosed in symptomatic neonates and even asymptomatic or mildly symptomatic children, teens, and adults.
Prenatal Diagnosis
Prenatal investigations by second-trimester two-dimensional (2D) ultrasound and/or MRI detect more than 60% of affected fetuses with right-sided defects and more than 80% of those with left-sided CDH [Beaumier et al 2015]. Ultrasound demonstrates herniated viscera with or without liver in the fetal thorax, absence of the normal position of the stomach bubble below the diaphragm, and mediastinal shift. MRI is especially useful for the prenatal diagnosis of thoracic lesions that are atypical or complicated by multiple abnormalities and for assessing lung volumes [Matsuoka et al 2003]. Estimates of fetal lung volume and descriptions of the amount of herniated viscera are often used as prognostic indicators (see Prognosis).
Color flow Doppler can be used to:
- Demonstrate abnormal positioning of the umbilical and portal veins, which are indicative of liver herniation;
- Identify right-sided hernias, which can be difficult to detect on ultrasound examination because of the similar echogenicity of lung and liver.
Fetal MRI is increasingly being used to confirm the diagnosis of CDH during the second and third trimester as a complementary tool to ultrasound studies to better define fetal anatomy [Kasprian et al 2006]. The primary advantage of fetal MRI is the ability to measure total lung volume and allow a quantitative assessment of the liver mass herniated into the chest. A systematic review and meta-analysis of observational studies suggests that fetal MRI can be used to predict neonatal survival with some caveats [Mayer et al 2011]:
- MRI has limited benefit early in gestation.
- Lung measurements in individuals with CDH are not standardized.
- Survival rates and treatment protocols from different centers are not comparable.
Postnatal Diagnosis
CDH can be detected in a neonate by:
- Presence of a scaphoid abdomen, diminished breath sounds ipsilateral to the side of the hernia, and displacement of the heart sounds contralateral to the hernia.
- Chest radiograph shows visible bowel gas above the diaphragm accompanied by a mediastinal shift.
Rarely, older children or adults with inconspicuous congenital diaphragmatic defects can be suspected because of respiratory or gastrointestinal symptoms including chronic cough, recurrent pulmonary infections, pleural effusions, pneumonia, or dysphagia. Intestinal obstruction and volvulus may be presenting symptoms, as abnormalities in the usual intestinal rotation during fetal development are common. Finally, some individuals may be diagnosed incidentally by plain chest radiographs or other imaging procedures. In these individuals, the degree of respiratory symptoms is correlated with the degree of pulmonary hypoplasia and can be limited.
Clinical Manifestations
Respiratory Compromise
Infants with CDH often present in the neonatal period with severe respiratory distress, occasionally after a stable period of 24-48 hours followed by acute respiratory distress. Breath sounds are diminished ipsilateral to the hernia. Almost all individuals with CDH have some degree of pulmonary hypoplasia. The pathogenesis of the pulmonary hypoplasia associated with CDH appears to have both a primary component (i.e., the hypoplasia occurs independent of the diaphragm defect) and a secondary component (i.e., arising from competition for thoracic space particularly in the lung ipsilateral to the hernia). Evidence for the presence of a primary defect in lung development arises mostly from studies in animal models, some of which show that the lung hypoplasia precedes the herniation of abdominal viscera.
Infants with CDH typically require mechanical ventilation and sometimes extracorporeal membrane oxygenation (ECMO) in the newborn period. Major respiratory complications include tracheobronchomalacia, pneumothorax, and secondary lung infection (especially viral pneumonia) that could precipitate terminal respiratory failure even months after surgery. Many infants require ongoing oxygen supplementation and diuretics following surgical correction of CDH. Given the remarkable growth and recuperative capacity of the lung, these treatments can usually be discontinued within the first two years of life.
By early childhood, few children have respiratory symptoms at rest; however, formal testing in older children shows small airway obstruction and diminished blood flow on ventilation-perfusion (V-Q) scan, especially to the lung ipsilateral to the hernia. Reduced exercise tolerance can be a lifelong problem. Intermittent wheezing requiring bronchodilator use is common in people with CDH, and they are at risk for respiratory decompensation with intercurrent illness.
Pulmonary Hypertension
Abnormal pulmonary vascular development and function is a significant problem in infants with CDH. Guidelines for the diagnosis of pediatric pulmonary hypertension were set forth by the American Heart Association and American Thoracic Society [Abman et al 2015]; however, individuals with CDH present a unique challenge because of the very young age of onset in PH. The mechanism of pulmonary hypertension in CDH is not completely understood. The size of the pulmonary vascular bed is decreased in the hypoplastic lungs and the vasculature displays medial and adventitial thickening [reviewed in Harting 2017]. Notable is the premature differentiation of smooth muscle cells into a contractile phenotype [Sluiter et al 2013]. Failure of relaxation of the pulmonary vasculature at birth is a critical component leading to pulmonary hypertension in CDH and it is the consequence of abnormal expression of both vasoconstrictors and vasodilators in distinct but converging pathways [McCulley et al 2018].
Gastrointestinal
"Failure to thrive" with growth parameters lower than than the third centile of normal is common among infants with more significant pulmonary hypoplasia and/or a more prolonged hospitalization following surgical repair of CDH. Growth failure is caused, in large part, by oral aversion and feeding difficulties (often requiring gastrostomy tube insertion for the first few years of life) and gastroesophageal reflux (frequently requiring pharmacotherapy and/or surgical fundoplication). Some infants and children require long-term high-calorie nutritional supplements.
Neurologic/Developmental
Reporting of neurodevelopmental outcomes is complicated by lack of standardization in terms of outcomes assessed, age of assessment, and metrics used (reviewed in IJsselstijn et al [2018]). The main independent predictor of intellectual disability is ECMO treatment. Mild (44%) or severe (13%) delay in at least one domain at age one year has been reported. Later, intelligence is reported in the normal range for the majority of individuals with isolated CDH, but educational support in school is often warranted and behavioral issues such as struggling with concentration and attention are common [Friedman et al 2008, Peetsold et al 2009, IJsselstijn et al 2018]. Preschool motor development scores in individuals with CDH are normal or mildly reduced, and improve with age; however, nonfocal neurologic abnormalities such as hypotonia are common, as are motor problems, especially in ECMO survivors [Nijhuis-van der Sanden et al 2009]. Nonspecific findings such as cortical atrophy, ventriculomegaly, and intracranial hemorrhage can be seen on neuroimaging studies [Ahmad et al 1999, Bouman et al 2000, Rasheed et al 2001].
Musculoskeletal
Chest asymmetry is found in as many as half of individuals with CDH. Pectus deformity, most often of the excavatum type, and scoliosis (≥10° Cobb's angle) are found in approximately 25% of individuals. These musculoskeletal abnormalities occur more often following repair of large diaphragmatic defects, possibly as a result of the extra tension exerted on the chest wall during surgical repair.
Sensorineural Hearing Loss
Sensorineural hearing loss (SNHL) has been found in 25% of individuals with CDH and as many as 100% of individuals treated with ECMO in some series [Rasheed et al 2001, Robertson et al 2002]. The fact that SNHL is late-onset and progressive, and therefore is not present on neonatal hearing screening, makes it difficult to compare studies and provide a more precise frequency. One study of ECMO survivors showed that SNHL was 2.5 times more common among those requiring ECMO for CDH than for other indications; however, other studies report similar frequencies after ECMO independent of the underlying diagnosis [van den Hondel et al 2013]. Other factors such as prolonged treatment with aminoglycosides, use of nitric oxide, prolonged or high-frequency mechanical ventilation, and/or metabolic alkalosis could also contribute to the development of SNHL [Fligor et al 2005].
Prognosis
Mortality. Mortality estimates range from 20% to 60% due to variation in patient populations and data collection techniques. Data from neonatal or referral centers, primarily including those with isolated left-sided Bochdalek hernia, report 80%-90% survival [Downard et al 2003]. However, population-based studies of outcome for all prenatally diagnosed individuals with CDH report mortality of at least 50%, if pregnancy terminations are included [Colvin et al 2005]. In a meta-analysis, Stege et al [2003] observed that approximately one quarter of all prenatally diagnosed fetuses were electively terminated, 3% spontaneously miscarried, and 3% were stillborn; 31% of the live-born infants died, the majority within the first 24 hours of life.
Prognostic indicators
- Whether the CDH is isolated or complex. Higher mortality occurs with complex CDH associated with a chromosome abnormality, a single-gene disorder, and/or the coexistence of major malformations. The presence of a cardiovascular malformation also indicates a worse prognosis [Cohen et al 2002].
- The size of the diaphragm defect [Lally et al 2007]
- The degree of pulmonary hypoplasia. Several methods are used to estimate the severity of pulmonary hypoplasia using imaging techniques. Prenatal estimates of lung-to-head ratio (LHR), observed/expected-total fetal lung volume (O/E-TFLV), and percent liver herniation (%LH) have been proposed as outcome predictors. The O/E-TFLV has the advantage of correcting for gestational age: LHR values increase as gestation progresses [Schaible et al 2012]. Using multivariate regression, MRI-based O/E-TFLV was shown to be an independent predictor of six-month mortality [Akinkuotu et al 2016].
- Liver herniation was associated with poorer prognosis in a large systematic review and meta-analysis of cases with prenatal detection of CDH [Mullassery et al 2010].
- The severity of pulmonary hypertension in the perinatal period. Pulmonary hypertension, which may progress to a late or chronic phase, is often not responsive to medical therapy [Kinsella et al 2005].
- Whether the hernia is right-sided, left-sided, or bilateral. Some, but not all, studies show that a right-sided hernia is associated with greater mortality than a left-sided hernia [Skari et al 2000]. Bilateral CDH always confers a high mortality. More recent studies suggest that fetuses with right-sided defects showed higher rates of adverse ultrasound predictors, but equivalent survival to left-sided defects [Sperling et al 2018].
Differential Diagnosis
Bronchogenic (foregut duplication) cysts result from abnormal budding of the ventral foregut [Knudtson & Grewal 2004]. They contain several components of the bronchi, including respiratory epithelia, mucous glands, and cartilage and may occur anywhere along the length of the trachea or esophagus. Most are diagnosed incidentally, although cysts can become infected or if large enough, can compress the esophagus and/or trachea.
Congenital cystic adenomatoid malformation (CCAM) is a developmental abnormality of the lung resulting from abnormal cell proliferation and decreased programmed cell death of lung tissue. Abnormally formed bronchi connect to the CCAM. Type I CCAM is most common and is distinguished by relatively large cysts and mucin production. Symptoms can result when CCAMs grow in size and compress structures in the mediastinum.
Cystic teratomas are benign tumors most often found in the anterior mediastinum [Jaggers & Balsara 2004]. They consist of several differentiated cell types derived from endoderm, ectoderm, and/or mesoderm. Cystic teratomas of the mediastinum are uncommon, comprising fewer than 10% of all tumors in that region.
Neurogenic tumors are the most common lesion found in the posterior mediastinum. They are likely to be of neural crest origin; the majority are benign. Examples include: neurilemoma, neurofibroma, ganglioneuroma, pheochromocytoma, and neuroblastoma. CT and MRI are helpful in establishing the diagnosis.
Paraesophageal hernia occurs when a portion of the stomach and sometimes part of the peritoneal sac containing the spleen or colon move into the chest cavity through the (normally occurring but) generally enlarged or dilated esophageal hiatus. More accurately, paraesophageal hernias are a type of hiatal hernia, in which the stomach gets "stuck" in the chest, rather than sliding back and forth between the thorax and abdomen. Approximately 5%-10% of (acquired) hernias are paraesophageal. They are rare in infancy and most commonly present in older adults.
Pulmonary agenesis refers to partial or complete absence of lung tissue that is caused by failure of lung bud development. It is often associated with additional congenital malformations.
Pulmonary sequestration results from primitive lung tissue that is not connected to the tracheobronchial tree. Sequestration may be intrapulmonary, occurring within the pleura of the normal lung, or extrapulmonary, occurring outside the normal lung within its own pleural sac. Extrapulmonary sequestration appears to arise from an accessory lung bud and often has associated anomalies, such as CDH. The most common presenting symptom is recurrent chest infections.
2. Genetic Causes of Congenital Diaphragmatic Hernia
Older data using standard cytogenetic and molecular cytogenetic techniques showed that approximately 15%-20% of individuals with CDH were identified as having a genetic cause for their diaphragm defect [Pober et al 2005, Pober 2008]. These frequencies are likely an underestimate and are evolving with the availability of next-generation sequencing and the identification of new CDH-associated genes. Genetic causes can be subdivided into monogenic causes (Table 1) and chromosome anomalies (Table 3). Though monogenic CDH can be isolated, it is more commonly syndromic.
Table 1.
Congenital Diaphragmatic Hernia: Monogenic Causes and Distinguishing Clinical Features
Table 2.
Congenital Diaphragmatic Hernia: Syndromic Causes of Unknown Genetic Etiology and Distinguishing Clinical Features
Chromosomal Causes of Congenital Diaphragmatic Hernia
Historically, cytogenetically detectable chromosome anomalies have been reported in approximately 10% of individuals with CDH (see Table 3). More recently with the advent of higher-resolution molecular cytogenetic technologies, additional copy number variations have been described in individuals with CDH.
Table 3.
Chromosome Anomalies Associated with Congenital Diaphragmatic Hernia
3. Evaluation Strategies to Identify the Genetic Cause of Congenital Diaphragmatic Hernia in a Proband
Establishing a specific genetic cause of congenital diaphragmatic hernia:
- Can aid in discussions of prognosis (which are beyond the scope of this GeneReview) and genetic counseling;
- Usually involves a medical history, physical examination, family history, imaging, and genomic/genetic testing;
- Is complicated by the extreme genetic heterogeneity of the condition, which requires a tiered approach to genetic testing.
Prenatal Evaluation Strategy
Medical history. CDH is commonly diagnosed prenatally by detecting the abnormal positioning of the stomach, liver, or other viscera in the fetal thorax. Polyhydramnios is frequently present. Additional congenital anomalies identified on the fetal ultrasound may direct the diagnosis to a specific cause of congenital diaphragmatic hernia. Posterolateral (Bochdalek) hernias most commonly present within hours after birth with signs of pulmonary distress. Tables 1, 2, and 3 provide a list of disorders frequently associated with CDH. Specific diagnostic investigations may be warranted when one of these conditions is suspected.
Physical examination. In addition to viable pregnancies, possible outcomes of pregnancies with CDH include miscarriage, fetal death, and termination of pregnancy. If the cause of the diaphragmatic defect is unknown, a fetopsy or autopsy examination that includes photographs and skeletal radiographs may identify co-occurring anomalies. At the time of autopsy, a sample for genomic/genetic testing may be obtained.
Family history. A three-generation family history should be taken, with attention to relatives with manifestations of congenital diaphragmatic hernia and/or infants who died in the perinatal period. Documentation of relevant findings in relatives should be obtained through direct examination or review of medical records, including results of molecular genetic testing.
Testing
Cytogenetic testing. Chorionic villus sampling (CVS) or amniocentesis for chromosome analysis (G-banded or Q-banded karyotype) are offered in many institutions as a first-line test in all fetuses prenatally diagnosed with CDH. Special attention must be paid in certain circumstances when mosaicism is suspected. Congenital diaphragmatic hernia, congenital heart defects, and rhizomelic limb shortening identified on ultrasound raise the suspicion for Pallister-Killian syndrome (PKS; see Table 3); however, the most frequent ultrasound finding in this condition is the combination of polyhydramnios, macrosomia, and limb shortening [Salzano et al 2018]. The isochromosome 12p characteristic of PKS may be missed in 50% of CVS samples, while the detection rate of amniocentesis is close to 90% [Salzano et al 2018].
Chromosomal microarray analysis (CMA) using oligonucleotide or SNP arrays to detect microdeletions and microduplications is indicated in a fetus with CDH and additional multiple anomalies and/or a suggestive family history, if the karyotype has not identified a diagnosis (see Table 3). Molecular cytogenetic testing may also be offered even in individuals with apparently isolated CDH, since complex or syndromic presentations can be difficult to appreciate on prenatal imaging.
Exome sequencing. To date, exome sequencing in the prenatal period is performed exclusively on a research basis in fetuses with abnormal ultrasound scans and normal microarray analysis of chorionic villi or amniocytes [Drury et al 2015]. Family trio exome sequencing is preferred in this situation, because of the reported high rate of de novo sequence variants in both isolated and complex CDH [Longoni et al 2017, Qi et al 2018].
For an introduction to exome sequencing click here. More detailed information for clinicians ordering genomic testing can be found here.
Targeted prenatal testing may be available in those situations in which the causative variant is known in an affected family member.
Postnatal Evaluation Strategy
Genetic testing should be directed by the clinical evidence garnered by the interpretation of medical history, physical examination, and family history.
Medical history. Respiratory distress is the most prominent finding in a newborn with CDH. The degree of pulmonary hypoplasia and pulmonary hypertension is not directly correlated with the genetic etiology of the condition; however, individuals with syndromic CDH and multiple anomalies usually have worse outcomes. Prenatal exposure to pharmacologic agents and environmental hazards (e.g., phenmetrazine, thalidomide, quinine, cadmium, lead, nitrofen) must be investigated and excluded [Stolar & Dillon 2012]. Newborns with CDH have an increased incidence of additional malformations that could assist in the differential diagnosis (see Tables 1, 2, and 3).
Physical examination. Infants with CDH should be evaluated by a clinical geneticist for other structural birth defects (e.g., craniofacial, ocular, cardiovascular, central nervous system, limb, genitourinary), as well as minor anomalies or dysmorphic features, that may suggest a specific genetic cause of CDH (see Tables 1, 2, and 3). In the absence of a specific diagnosis, permission should be sought to perform an autopsy on deceased infants with CDH, especially in the perinatal period. The autopsy should include photographs, skeletal radiographs, and a sample for molecular genetic testing. A skin biopsy for cell line development and banking in a repository could provide additional genetic material should new tests be developed in the future; however, this option may only be available within a research study.
Family history. A three-generation family history should be taken, with attention to relatives with manifestations of congenital diaphragmatic hernia and/or infants who died in the perinatal period and documentation of relevant findings through direct examination or review of medical records, including results of molecular genetic testing.
Testing
Molecular genetic testing approaches can also include a combination of comprehensive genomic testing (chromosomal microarray analysis, exome sequencing, genome sequencing), karyotype, and gene-targeted testing (single-gene testing, multigene panel).
Note: (1) In an otherwise healthy individual with isolated CHD and a negative family history, currently available clinical genetic testing has a relatively low yield. However, the decision to proceed with testing should be made by an experienced geneticist on a case-by-case basis and should be regularly reevaluated in light of the individual’s developmental course, as well as advances in identifying genetic contributions to CDH. (2) Special attention must be paid when considering the diagnosis of isochromosome 12p (Pallister-Killian syndrome; see Table 3) since peripheral blood chromosome studies are almost invariably normal in the postnatal period. A recent report, however, demonstrates presence of iso 12p mosaicism detected by CMA performed on DNA extracted from peripheral blood [Theisen et al 2009].
Chromosomal microarray analysis (CMA) using oligonucleotide or SNP arrays to detect microdeletions and microduplications should be considered as first-line testing in a child who has CDH with additional multiple anomalies and/or a suggestive family history (see Table 3). CMA is particularly useful in individuals with multiple anomalies whose constellation is not suggestive of a specific disorder.
Standard cytogenetic testing has specialized utility in the postnatal period. If an aneuploidy (e.g., trisomy 21) or a chromosome rearrangement is suspected postnatally in an infant with CDH, a karyotype may be considered prior to performing CMA; see Table 3 and Note: (2) above.
Serial single-gene testing can be considered if clinical findings and/or family history indicate that pathogenic variants in a particular gene are most likely (see Table 1). Because it is laborious and frequently inconclusive, serial single-gene testing is not frequently employed and comprehensive genomic testing is preferred in its stead in the absence of a strong clinical suspicion.
A multigene panel that includes some or all of the genes listed in the Table 1 is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. No laboratories listed in the NCBI Genetic Testing Registry (GTR) (last accession: February 1, 2019) offer multigene panels specifically designed around diaphragmatic defects, although some CDH-associated genes are frequently included in congenital heart defect panels. However, these multigene panels are of limited utility for individuals with nonsyndromic CDH and narrow utility for those with syndromic CDH. Clinical exome sequencing should be considered if comprehensive testing is clinically indicated.
Comprehensive genomic testing (which does not require the clinician to a priori hypothesize which gene[s] are likely involved) may be considered in individuals with negative prior testing, or as first-line testing in individuals for whom a hypothesis for a single-gene disorder cannot be made readily. Exome sequencing is most commonly used; genome sequencing is also possible.
Exome sequencing. Clinical exome sequencing in the postnatal period is recommended as second- or third-line testing due to considerations of cost and reimbursement (exome sequencing may also be offered as part of certain research studies if clinically unavailable). Its utility is presently confined to those with multiple anomalies and negative CMA and karyotype results. When exome sequencing is performed, concurrent or sequential trio testing is preferred to proband-only testing because of the reported high rate of de novo sequence variants in both isolated and complex CDH.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
4. Management
Treatment of Manifestations
Proper management of newborns with CDH must start in the delivery room; recently, investigative therapies may start in the prenatal period. A standardized protocol for neonatal management and treatment of infants with CDH was proposed by the CDH EURO Consortium Consensus [Reiss et al 2010], and more recently updated by Snoek et al [2016].
Respiratory support. Emphasis must be on preventing secondary lung injury. Newborns should be intubated immediately in the delivery room to avoid bag-mask ventilation and inflation of bowel that has herniated into the chest. Care should be taken not to induce barotrauma from bag ventilation before the neonate can be transitioned to an appropriate ventilator. In the 1990s, it was recognized that minimizing aggressive ventilation by limiting ventilator settings and/or allowing the neonate to do some of the breathing work resulted in improved outcomes. It was also recognized that infants did not need to be rushed to surgery and benefited from stabilization of respiratory and cardiovascular status prior to diaphragmatic repair. Assuring end-organ perfusion and stability instead of perfect physiologic values or immediate anatomic correction is now the standard of care [Wung et al 1995, Boloker et al 2002, Mohseni-Bod & Bohn 2007].
Extracorporeal membrane oxygenation (ECMO) continues to be used frequently in tertiary centers as a rescue therapy for neonates with critical cardiopulmonary deterioration. Critical factors to consider before initiating ECMO are the presence of coexisting anomalies (lethal conditions in particular), gestational age, birth weight, uncontrollable bleeding or uncorrectable bleeding diathesis, intracranial hemorrhage, and outlook of medical management [Garcia et al 2014, Cairo et al 2018]. The clinical utility of ECMO in improving overall survival hinges on the selection of the population most likely to benefit [discussed in Kays 2017].
Complications of ECMO treatment include air embolism, neurologic complications (e.g., intracranial hemorrhage, infarct, and seizures), cannulation site bleeding, coagulation abnormalities including disseminated intravascular coagulation, left-to-right shunting through the patent ductus arteriosus due to rapid and dramatic decrease in pulmonary hypertension, kidney failure, systemic hypertension, and infection [Garcia et al 2014]. Due to the complex nature of treatment and the severity of possible complications, significant experience and a multidisciplinary team are required to manage individuals on ECMO.
Pulmonary hypertension. Unfortunately, therapies that have been extremely successful in treating the usual persistent pulmonary hypertension of the newborn (PPHN) have not been widely successful in attenuating CDH-associated pulmonary hypertension and right ventricular failure. Evidence and recommendations for the diagnosis, monitoring, and management of pulmonary hypertension are reviewed in the CDH EURO Consortium Consensus – 2015 update [Snoek et al 2016]. Since some infants respond to nitric oxide (NO) [Okuyama et al 2002], a trial of NO with careful documentation of response with echocardiography prior to continuation is warranted. Phosphodiesterase inhibitors have been used in some individuals [Mohseni-Bod & Bohn 2007, Noori et al 2007]. Other therapies that have been introduced in the acute neonatal treatment phase for CDH but are controversial include the use of surfactant and perflubron [Fauza et al 2001, Hirschl et al 2003]. Unfortunately, the lack of large randomized controlled trials makes it difficult to determine which of these treatments may be beneficial. The presence of surfactant deficiency in those with CDH may depend on the sub-population, thus making it difficult to determine whether exogenous surfactant would be efficacious in large trials [Jani et al 2009, Janssen et al 2009].
Surveillance
Since both pre- and postnatal advances in treatment have increased survival of high-risk individuals, it is important to provide close follow up and support for major complications and potential long-term morbidities. Long-term follow up for infants with CDH is ideally provided at a specialized center by a multidisciplinary team consisting of a pediatric surgeon, surgical nurse specialist, cardiologist, nutritionist, pulmonologist, and developmental pediatrician. This type of team can recognize, treat, and coordinate care for the many medical complications frequently found in long-term survivors with CDH.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Prenatal treatment. Treatment of severely affected infants has shifted to the prenatal period, as rescue of severe pulmonary hypoplasia is possible if done at the correct time. The discovery that laryngeal obstruction leads to lung distention from retained fluid prompted tracheal occlusion studies in animal models and in humans [Lipshutz et al 1997, Harrison et al 2003]. In one US randomized trial of fetal endoscopic tracheal occlusion, the treatment group experienced a high rate of preterm delivery and did not have improved morbidity or mortality rates [Harrison et al 2003]. However, since that time, advances have been made in tracheal occlusion techniques and in predicting which fetuses would most likely die without any intervention. Currently, high-risk fetuses in Europe or in a few US centers may receive tracheal occlusion by fetal endoscopic balloon placement. The procedure is performed in the second trimester when the observed/expected lung-area to head-circumference ratios are ≤1.0. Generally, fetuses selected for this procedure have isolated CDH (as well as can be determined by prenatal imaging) without chromosome aberrations. Although the treatment remains investigational, initial survival rates in high-risk fetuses are increased [Araujo Júnior et al 2017].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
5. Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Congenital diaphragmatic hernia (CDH) may occur as an isolated finding, as part of a genetic syndrome or chromosome abnormality, or as part of a complex but nonsyndromic set of findings.
Kindreds representing both syndromic and nonsyndromic CDH consistent with autosomal dominant, autosomal recessive, and X-linked patterns of inheritance have been reported. Some published pedigrees suggest reduced penetrance and variable expressivity in individuals with heterozygous pathogenic variants associated with autosomal dominant CDH [Yu et al 2013, Longoni et al 2015].
Risks to Family Members of a Proband With an Identified Genetic Etiology
If a proband is found to have a specific genetic disorder, syndrome, or association with CDH (see Table 1) or an inherited or de novo chromosome abnormality (see Table 3), recurrence risk depends on the mode of inheritance associated with the condition, and genetic counseling for that condition is indicated.
Empiric Risks to Family Members of a Proband Without an Identified Genetic Etiology
Isolated CDH
- The majority of individuals with isolated CDH represent simplex cases (i.e., the only affected member of the family); however, a few families are multiplex (i.e., ≥2 relatives have isolated CDH).
- The estimated risk for recurrence of isolated Bochdalek CDH in a sib is less than 2% [Pober et al 2005]. These studies primarily assessed the frequency of sibs diagnosed with CDH who were born prior to the proband (precurrence); there is no systematic or consecutive series on sib recurrence to date.
Complex CDH. Counseling for individuals with complex CDH in whom a specific genetic disorder is not recognized is problematic:
- Some cases of complex CDH are probably caused by dominant de novo pathogenic variants, and therefore pose a low recurrence risk to the sibs of the proband.
- Some are probably unrecognized syndromic conditions.
- Some may be multifactorial disorders with a low recurrence risk.
- Non-genetic causes including stochastic events, epigenetic modifications, or teratogenic/environmental exposures are possible as well.
Thus, counseling in this setting should be as for other multiple congenital anomaly disorders of unknown etiology. Specifically, the estimated recurrence risk to sibs is "low," but this estimate represents an averaging of a negligible, or very low, recurrence risk in the majority of families together with a higher recurrence risk (as high as 25%-50%) in the minority of families.
Related Genetic Counseling Issues
Offspring of a proband. A growing number of CDH survivors are reaching their reproductive years due to improved surgical techniques and medical management. At this time, no studies have been conducted to interrogate systematically the recurrence risk in this population. However, in theory, the risk of having a child with CDH could be as high as 50% in an individual with a de novo pathogenic variant or an inherited autosomal dominant pathogenic variant, assuming complete penetrance.
Variable expressivity. The relationship between Bochdalek hernias and muscle migration defects (eventrations) is unknown, but they may be related entities. Both abnormalities have been reported as occurring in different members of the same family and in the same individual, as reported in Ackerman et al [2005] and in individuals with pathogenic variants in the same gene [Jordan et al 2018].
Among multiplex families with CDH, concordance is extremely high among affected relatives both for the specific type and the side of the diaphragm defect, although the size of the defect can vary. Very occasionally, one affected sib has unilateral CDH while a second affected sib has bilateral CDH, or one sib has an eventration while a second has a diaphragmatic hernia. Also, occasionally, one sib has CDH only while a second affected sib has CDH plus another common birth defect such as a cardiovascular malformation or polydactyly. Whether these latter cases represent differing manifestations of a monogenic condition influenced by genetic modifiers, environmental modifiers, or stochastic fluctuations, or even multifactorial inheritance is not yet known.
Discordant monozygotic twins. Most monozygotic twin pairs in consecutive series are discordant (i.e., one member of the twin pair has CDH while the other twin does not) [Pober et al 2005, Veenma et al 2012, Wang et al 2019]. The reason for such high rates of phenotype discordance is presently unknown and a variety of mechanisms, both genetic and non-genetic, could account for these findings. Small cohort studies in discordant monozygotic twins showed no evidence of pathogenic copy number variant discordance [Veenma et al 2012] or sequence variant discordance [Zhang et al 2016].
Prenatal Testing and Preimplantation Genetic Testing
Low-risk pregnancies (i.e., those without a known family history of CDH). The majority of probands with CDH are diagnosed prenatally by ultrasound examination, which demonstrates herniated viscera with or without liver in the fetal thorax, absence of the normal position of the stomach bubble below the diaphragm, and mediastinal shift [Stege et al 2003, Tonks et al 2004]. Although not specific for CDH, polyhydramnios is often detected [Witters et al 2001].
When CDH is found on routine prenatal ultrasound examination, both a high-resolution ultrasound examination and fetal MRI to determine the presence of additional structural anomalies are indicated. Chromosome analysis of fetal cells obtained by amniocentesis should be considered in all pregnancies while CMA should strongly be considered when CDH is present in conjunction with additional anomalies. (See Evaluation Strategies to Identify the Genetic Cause of Congenital Diaphragmatic Hernia in a Proband for additional details.)
All fetuses with CDH should be evaluated for the presence of syndromes and/or additional major malformations given that they so commonly coexist and significantly affect the prognosis. Involvement of a clinical geneticist in the evaluation of these families can be helpful. The measurement of either the expected/observed LHR or the lung volume by fetal MRI have been useful to predict outcome; however, since the predictive value of these measurements varies from center to center, results must be interpreted with caution.
High-risk pregnancies (i.e., those with a family history of CDH). Consultation in a high-risk obstetric center should be offered.
If the genetic cause of CDH has been identified in an affected family member, prenatal testing and preimplantation genetic testing are possible. Counseling should include a discussion of possible reduced penetrance in autosomal dominant forms of CDH [Yu et al 2013, Longoni et al 2015].
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- CDH InternationalEmail: info@cdhi.org
- CDH UKUnited KingdomPhone: 0800 731 6991Email: committee@cdhuk.org.uk
- Medical Home Portal
- MedlinePlus
- National Organization for Rare Disorders
- Compassionate FriendsSupporting Family After a Child DiesPhone: 877-969-0010
- SHARE Pregnancy and Infant Loss SupportPhone: 800-821-6819Email: info@nationalshare.org
Chapter Notes
Author History
Kate Guernsey Ackerman, MD; University of Rochester (2006-2019)
Frances A High, MD, PhD (2019-present)
Mauro Longoni, MD (2019-present)
Barbara R Pober, MD (2006-present)
Meaghan K Russell, MPH; MassGeneral Hospital for Children (2006-2019)
Revision History
- 5 November 2020 (aa/sw) Revision: added PIGN-related Fryns syndrome; removed MCAHS (not associated with congenital diaphragmatic hernia)
- 28 March 2019 (sw) Comprehensive update posted live
- 16 March 2010 (me) Comprehensive update posted live
- 1 February 2006 (me) Review posted live
- 6 June 2005 (brp) Original submission
References
Literature Cited
- Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, Hanna BD, Rosenzweig EB, Raj JU, Cornfield D, Stenmark KR, Steinhorn R, Thébaud B, Fineman JR, Kuehne T, Feinstein JA, Friedberg MK, Earing M, Barst RJ, Keller RL, Kinsella JP, Mullen M, Deterding R, Kulik T, Mallory G, Humpl T, Wessel DL, et al. Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation. 2015;132:2037–99. [PubMed: 26534956]
- Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, Rao C, Pober BR, Babiuk RP, Epstein JA, Greer JJ, Beier DR. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. 2005;1:58–65. [PMC free article: PMC1183529] [PubMed: 16103912]
- Ahmad A, Gangitano E, Odell RM, Doran R, Durand M. Survival, intracranial lesions, and neurodevelopmental outcome in infants with congenital diaphragmatic hernia treated with extracorporeal membrane oxygenation. J Perinatol. 1999;19:436–40. [PubMed: 10685274]
- Akinkuotu AC, Cruz SM, Abbas PI, Lee TC, Welty SE, Olutoye OO, Cassady CI, Mehollin-Ray AR, Ruano R, Belfort MA, Cass DL. Risk-stratification of severity for infants with CDH: Prenatal versus postnatal predictors of outcome. J Pediatr Surg. 2016;51:44–8. [PubMed: 26563530]
- Araujo Júnior E, Tonni G, Martins WP, Ruano R. Procedure-related complications and survival following fetoscopic endotracheal occlusion (FETO) for severe congenital diaphragmatic hernia: systematic review and meta-analysis in the FETO era. Eur J Pediatr Surg. 2017;27:297–305. [PubMed: 27522127]
- Bagłaj M, Dorobisz U. Late-presenting congenital diaphragmatic hernia in children: a literature review. Pediatr Radiol. 2005;35:478–88. [PubMed: 15778858]
- Beaumier CK, Beres AL, Puligandla PS, Skarsgard ED, et al. Clinical characteristics and outcomes of patients with right congenital diaphragmatic hernia: A population-based study. J Pediatr Surg. 2015;50:731–3. [PubMed: 25783377]
- Boloker J, Bateman DA, Wung JT, Stolar CJ. Congenital diaphragmatic hernia in 120 infants treated consecutively with permissive hypercapnea/spontaneous respiration/elective repair. J Pediatr Surg. 2002;37:357–66. [PubMed: 11877648]
- Bouman NH, Koot HM, Tibboel D, Hazebroek FW. Children with congenital diaphragmatic hernia are at risk for lower levels of cognitive functioning and increased emotional and behavioral problems. Eur J Pediatr Surg. 2000;10:3–7. [PubMed: 10770239]
- Burgos CM, Frenckner B. Addressing the hidden mortality in CDH: A population-based study. J Pediatr Surg. 2017;52:522–5. [PubMed: 27745705]
- Cairo SB, Arbuthnot M, Boomer L, Dingeldein MW, Feliz A, Gadepalli S, Newton CR, Puligandla P, Ricca R Jr, Rycus P, Vogel AM, Yu G, Chen Z, Rothstein DH, et al. Comparing percutaneous to open access for extracorporeal membrane oxygenation in pediatric respiratory failure. Pediatr Crit Care Med. 2018;19:981–91. [PMC free article: PMC6173194] [PubMed: 30080776]
- Cohen MS, Rychik J, Bush DM, Tian ZY, Howell LJ, Adzick NS, Flake AW, Johnson MP, Spray TL, Crombleholme TM. Influence of congenital heart disease on survival in children with congenital diaphragmatic hernia. J Pediatr. 2002;141:25–30. [PubMed: 12091847]
- Colvin J, Bower C, Dickinson JE, Sokol J. Outcomes of congenital diaphragmatic hernia: a population-based study in Western Australia. Pediatrics. 2005;116:e356–63. [PubMed: 16140678]
- Downard CD, Jaksic T, Garza JJ, Dzakovic A, Nemes L, Jennings RW, Wilson JM. Analysis of an improved survival rate for congenital diaphragmatic hernia. J Pediatr Surg. 2003;38:729–32. [PubMed: 12720181]
- Drury S, Williams H, Trump N, Boustred C, Lench N, Scott RH, Chitty LS, et al. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat Diagn. 2015;35:1010–7. [PubMed: 26275891]
- Fauza DO, Hirschl RB, Wilson JM. Continuous intrapulmonary distension with perfluorocarbon accelerates lung growth in infants with congenital diaphragmatic hernia: initial experience. J Pediatr Surg. 2001;36:1237–40. [PubMed: 11479865]
- Fligor BJ, Neault MW, Mullen CH, Feldman HA, Jones DT. Factors associated with sensorineural hearing loss among survivors of extracorporeal membrane oxygenation therapy. Pediatrics. 2005;115:1519–28. [PubMed: 15930212]
- Friedman S, Chen C, Chapman J, Jeruss S, Terrin N, Tighiouart H, Parsons S, Wilson J. Neurodevelopmental outcomes of congenital diaphragmatic hernia survivors in a multidisciplinary clinic at ages 1 and 3. J Pediatr Surg. 2008;43:1035–43. [PubMed: 18558179]
- Garcia AV, Thirumoorthi AS, Stolar CJH. Extracorporeal membrane oxygenation. In: Ashcraft KW, Holcomb GW III, Murphy JP, Ostlie DJ. Ashcraft's Pediatric Surgery. 2014:80-93.
- Harrison MR, Keller RL, Hawgood SB, Kitterman JA, Sandberg PL, Farmer DL, Lee H, Filly RA, Farrell JA, Albanese CT. A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia. N Engl J Med. 2003;349:1916–24. [PubMed: 14614166]
- Harting MT. Congenital diaphragmatic hernia-associated pulmonary hypertension. Semin Pediatr Surg. 2017;26:147–53. [PubMed: 28641752]
- Hirschl RB, Philip WF, Glick L, Greenspan J, Smith K, Thompson A, Wilson J, Adzick NS. A prospective, randomized pilot trial of perfluorocarbon-induced lung growth in newborns with congenital diaphragmatic hernia. J Pediatr Surg. 2003;38:283–9. [PubMed: 12632336]
- IJsselstijn H, Breatnach C, Hoskote A, Greenough A, Patel N, Capolupo I, Morini F, Scharbatke H, Kipfmueller F, Ertresvag K, Kraemer U, Braguglia A, Wessel L, van Heijst AFJ, Moinichen I, Emblem R, Tibboel D, et al. Defining outcomes following congenital diaphragmatic hernia using standardised clinical assessment and management plan (SCAMP) methodology within the CDH EURO consortium. Pediatr Res. 2018;84:181–9. [PubMed: 29915407]
- Jaggers J, Balsara K. Mediastinal masses in children. Semin Thorac Cardiovasc Surg. 2004;16:201–8. [PubMed: 15619186]
- Jani JC, Nicolaides KH, Gratacós E, Valencia CM, Doné E, Martinez JM, Gucciardo L, Cruz R, Deprest JA. Severe diaphragmatic hernia treated by fetal endoscopic tracheal occlusion. Ultrasound Obstet Gynecol. 2009;34:304–10. [PubMed: 19658113]
- Janssen DJ, Zimmermann LJ, Cogo P, Hamvas A, Bohlin K, Luijendijk IH, Wattimena D, Carnielli VP, Tibboel D. Decreased surfactant phosphatidylcholine synthesis in neonates with congenital diaphragmatic hernia during extracorporeal membrane oxygenation. Intensive Care Med. 2009;35:1754–60. [PMC free article: PMC2749174] [PubMed: 19582395]
- Jordan VK, Beck TF, Hernandez-Garcia A, Kundert PN, Kim BJ, Jhangiani SN, Gambin T, Starkovich M, Punetha J, Paine IS, Posey JE, Li AH, Muzny D, Hsu CW, Lashua AJ, Sun X, Fernandes CJ, Dickinson ME, Lally KP, Gibbs RA, Boerwinkle E, Lupski JR, Scott DA. The role of FREM2 and FRAS1 in the development of congenital diaphragmatic hernia. Hum Mol Genet. 2018;27:2064–75. [PMC free article: PMC5985720] [PubMed: 29618029]
- Kasprian G, Balassy C, Brugger PC, Prayer D. MRI of normal and pathological fetal lung development. Eur J Radiol. 2006;57:261–70. [PubMed: 16413987]
- Kays DW. ECMO in CDH: Is there a role? Semin Pediatr Surg. 2017;26:166–70. [PubMed: 28641755]
- Kinsella JP, Ivy DD, Abman SH. Pulmonary vasodilator therapy in congenital diaphragmatic hernia: acute, late, and chronic pulmonary hypertension. Semin Perinatol. 2005;29:123–8. [PubMed: 16052736]
- Knudtson J, Grewal H. Thoracoscopic excision of a paraesophageal bronchogenic cyst in a child. JSLS. 2004;8:179–82. [PMC free article: PMC3015520] [PubMed: 15119666]
- Lally KP, Lally PA, Lasky RE, Tibboel D, Jaksic T, Wilson JM, Frenckner B, Van Meurs KP, Bohn DJ, Davis CF, Hirschl RB. Defect size determines survival in infants with congenital diaphragmatic hernia. Pediatrics. 2007;120:e651–7. [PubMed: 17766505]
- Lipshutz GS, Albanese CT, Feldstein VA, Jennings RW, Housley HT, Beech R, Farrell JA, Harrison MR. Prospective analysis of lung-to-head ratio predicts survival for patients with prenatally diagnosed congenital diaphragmatic hernia. J Pediatr Surg. 1997;32:1634–6. [PubMed: 9396544]
- Longoni M, High FA, Qi H, Joy MP, Hila R, Coletti CM, Wynn J, Loscertales M, Shan L, Bult CJ, Wilson JM, Shen Y, Chung WK, Donahoe PK. Genome-wide enrichment of damaging de novo variants in patients with isolated and complex congenital diaphragmatic hernia. Hum Genet. 2017;136:679–91. [PMC free article: PMC5453716] [PubMed: 28303347]
- Longoni M, Russell MK, High FA, Darvishi K, Maalouf FI, Kashani A, Tracy AA, Coletti CM, Loscertales M, Lage K, Ackerman KG, Woods SA, Ward-Melver C, Andrews D, Lee C, Pober BR, Donahoe PK. Prevalence and penetrance of ZFPM2 mutations and deletions causing congenital diaphragmatic hernia. Clin Genet. 2015;87:362–7. [PMC free article: PMC4410767] [PubMed: 24702427]
- Matsuoka S, Takeuchi K, Yamanaka Y, Kaji Y, Sugimura K, Maruo T. Comparison of magnetic resonance imaging and ultrasonography in the prenatal diagnosis of congenital thoracic abnormalities. Fetal Diagn Ther. 2003;18:447–53. [PubMed: 14564118]
- Mayer S, Klaritsch P, Petersen S, Done E, Sandaite I, Till H, Claus F, Deprest JA. The correlation between lung volume and liver herniation measurements by fetal MRI in isolated congenital diaphragmatic hernia: a systematic review and meta-analysis of observational studies. Prenat Diagn. 2011;31:1086–96. [PubMed: 21915885]
- McCulley DJ, Wienhold MD, Hines EA, Hacker TA, Rogers A, Pewowaruk RJ, Zewdu R, Chesler NC, Selleri L, Sun X. PBX transcription factors drive pulmonary vascular adaptation to birth. J Clin Invest. 2018;128:655–67. [PMC free article: PMC5785269] [PubMed: 29251627]
- Merrell AJ, Ellis BJ, Fox ZD, Lawson JA, Weiss JA, Kardon G. Muscle connective tissue controls development of the diaphragm and is a source of congenital diaphragmatic hernias. Nat Genet. 2015;47:496–504. [PMC free article: PMC4414795] [PubMed: 25807280]
- Mohseni-Bod H, Bohn D. Pulmonary hypertension in congenital diaphragmatic hernia. Semin Pediatr Surg. 2007;16:126–33. [PubMed: 17462565]
- Mullassery D, Ba'ath ME, Jesudason EC, Losty PD. Value of liver herniation in prediction of outcome in fetal congenital diaphragmatic hernia: a systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2010;35:609–14. [PubMed: 20178116]
- Nijhuis-van der Sanden MW, van der Cammen-van Zijp MH, Janssen AJ, Reuser JJ, Mazer P, van Heijst AF, Gischler SJ, Tibboel D, Kollée LA. Motor performance in five-year-old extracorporeal membrane oxygenation survivors: a population-based study. Crit Care. 2009;13:R47. [PMC free article: PMC2689491] [PubMed: 19341476]
- Noori S, Friedlich P, Wong P, Garingo A, Seri I. Cardiovascular effects of sildenafil in neonates and infants with congenital diaphragmatic hernia and pulmonary hypertension. Neonatology. 2007;91:92–100. [PubMed: 17344658]
- Okuyama H, Kubota A, Oue T, Kuroda S, Ikegami R, Kamiyama M, Kitayama Y, Yagi M. Inhaled nitric oxide with early surgery improves the outcome of antenatally diagnosed congenital diaphragmatic hernia. J Pediatr Surg. 2002;37:1188–90. [PubMed: 12149699]
- Peetsold MG, Huisman J, Hofman VE, Heij HA, Raat H, Gemke RJ. Psychological outcome and quality of life in children born with congenital diaphragmatic hernia. Arch Dis Child. 2009;94:834–40. [PubMed: 19531530]
- Pober BR. Genetic aspects of human congenital diaphragmatic hernia. Clin Genet. 2008;74:1–15. [PMC free article: PMC2872786] [PubMed: 18510546]
- Pober BR, Lin A, Russell M, Ackerman KG, Chakravorty S, Strauss B, Westgate MN, Wilson J, Donahoe PK, Holmes LB. Infants with Bochdalek diaphragmatic hernia: sibling precurrence and monozygotic twin discordance in a hospital-based malformation surveillance program. Am J Med Genet A. 2005;138A:81–8. [PMC free article: PMC2891716] [PubMed: 16094667]
- Qi H, Yu L, Zhou X, Wynn J, Zhao H, Guo Y, Zhu N, Kitaygorodsky A, Hernan R, Aspelund G, Lim FY, Crombleholme T, Cusick R, Azarow K, Danko ME, Chung D, Warner BW, Mychaliska GB, Potoka D, Wagner AJ, ElFiky M, Wilson JM, Nickerson D, Bamshad M, High FA, Longoni M, Donahoe PK, Chung WK, Shen Y. De novo variants in congenital diaphragmatic hernia identify MYRF as a new syndrome and reveal genetic overlaps with other developmental disorders. PLoS Genet. 2018;14:e1007822. [PMC free article: PMC6301721] [PubMed: 30532227]
- Rasheed A, Tindall S, Cueny DL, Klein MD, Delaney-Black V. Neurodevelopmental outcome after congenital diaphragmatic hernia: Extracorporeal membrane oxygenation before and after surgery. J Pediatr Surg. 2001;36:539–44. [PubMed: 11283873]
- Reiss I, Schaible T, van den Hout L, Capolupo I, Allegaert K, van Heijst A, Gorett Silva M, Greenough A, Tibboel D, et al. Standardized postnatal management of infants with congenital diaphragmatic hernia in Europe: the CDH EURO Consortium consensus. Neonatology. 2010;98:354–64. [PubMed: 20980772]
- Robertson CM, Tyebkhan JM, Hagler ME, Cheung PY, Peliowski A, Etches PC. Late-onset, progressive sensorineural hearing loss after severe neonatal respiratory failure. Otol Neurotol. 2002;23:353–6. [PubMed: 11981395]
- Salzano E, Raible SE, Kaur M, Wilkens A, Sperti G, Tilton RK, Bettini LR, Rocca A, Cocchi G, Selicorni A, Conlin LK, McEldrew D, Gupta R, Thakur S, Izumi K, Krantz ID. Prenatal profile of Pallister-Killian syndrome: Retrospective analysis of 114 pregnancies, literature review and approach to prenatal diagnosis. Am J Med Genet A. 2018;176:2575–86. [PubMed: 30289601]
- Schaible T, Büsing KA, Felix JF, Hop WC, Zahn K, Wessel L, Siemer J, Neff KW, Tibboel D, Reiss I, van den Hout L. Prediction of chronic lung disease, survival and need for ECMO therapy in infants with congenital diaphragmatic hernia: additional value of fetal MRI measurements? Eur J Radiol. 2012;81:1076–82. [PubMed: 21458944]
- Shanmugam H, Brunelli L, Botto LD, Krikov S, Feldkamp ML. Epidemiology and prognosis of congenital diaphragmatic hernia: a population-based cohort study in Utah. Birth Defects Res. 2017;109:1451–9. [PubMed: 28925604]
- Skari H, Bjornland K, Haugen G, Egeland T, Emblem R. Congenital diaphragmatic hernia: a meta-analysis of mortality factors. J Pediatr Surg. 2000;35:1187–97. [PubMed: 10945692]
- Sluiter I, van der Horst I, van der Voorn P, Boerema-de Munck A, Buscop-van Kempen M, de Krijger R, Tibboel D, Reiss I, Rottier RJ. Premature differentiation of vascular smooth muscle cells in human congenital diaphragmatic hernia. Exp Mol Pathol. 2013;94:195–202. [PubMed: 23018129]
- Snoek KG, Reiss IK, Greenough A, Capolupo I, Urlesberger B, Wessel L, Storme L, Deprest J, Schaible T, van Heijst A, Tibboel D, et al. Standardized postnatal management of infants with congenital diaphragmatic hernia in Europe: the CDH EURO Consortium Consensus - 2015 update. Neonatology. 2016;110:66–74. [PubMed: 27077664]
- Sperling JD, Sparks TN, Berger VK, Farrell JA, Gosnell K, Keller RL, Norton ME, Gonzalez JM. Prenatal diagnosis of congenital diaphragmatic hernia: does laterality predict perinatal outcomes? Am J Perinatol. 2018;35:919–24. [PMC free article: PMC6033692] [PubMed: 29304545]
- Stege G, Fenton A, Jaffray B. Nihilism in the 1990s: the true mortality of congenital diaphragmatic hernia. Pediatrics. 2003;112:532–5. [PubMed: 12949279]
- Stolar CJH, Dillon PW. Congenital diaphragmatic hernia and eventration. In: Coran AG, Caldamone A, Adzick NS, Krummel TM, Laberge J-M, Shamberger R, eds. Pediatric Surgery. 7 ed. Chap 63. US Elsevier. 2012.
- Stoll C, Alembik Y, Dott B, Roth MP. Associated non diaphragmatic anomalies among cases with congenital diaphragmatic hernia. Genet Couns. 2015;26:281–98. [PubMed: 26625659]
- Theisen A, Rosenfeld J, Farrell S, Harris C, Wetzel H, Torchia B, Bejjani B, Ballif B, Shaffer L. aCGH detects partial tetrasomy of 12p in blood from Pallister-Killian syndrome cases without invasive skin biopsy. Am J Med Genet A. 2009;149A:914–8. [PubMed: 19353629]
- Tonks A, Wyldes M, Somerset DA, Dent K, Abhyankar A, Bagchi I, Lander A, Roberts E, Kilby MD. Congenital malformations of the diaphragm: findings of the West Midlands Congenital Anomaly Register 1995 to 2000. Prenat Diagn. 2004;24:596–604. [PubMed: 15305345]
- van den Hondel D, Madderom MJ, Goedegebure A, Gischler SJ, Mazer P, Tibboel D, Ijsselstijn H. Sensorineural hearing loss and language development following neonatal extracorporeal membrane oxygenation. Pediatr Crit Care Med. 2013;14:62–9. [PubMed: 23249782]
- Veenma D, Brosens E, de Jong E, van de Ven C, Meeussen C, Cohen-Overbeek T, Boter M, Eussen H, Douben H, Tibboel D, de Klein A. Copy number detection in discordant monozygotic twins of congenital diaphragmatic hernia (CDH) and esophageal atresia (EA) cohorts. Eur J Hum Genet. 2012;20:298–304. [PMC free article: PMC3283183] [PubMed: 22071887]
- Wang W, Pan W, Chen J, Xie W, Liu M, Wang J. Outcomes of congenital diaphragmatic hernia in one of the twins. Am J Perinatol. 2019;36:1304–9. [PubMed: 30609432]
- Wang Y, Hu J, Druschel CM, Kirby RS. Twenty-five-year survival of children with birth defects in New York State: a population-based study. Birth Defects Res A Clin Mol Teratol. 2011;91:995–1003. [PubMed: 21960515]
- Witters I, Legius E, Moerman P, Deprest J, Van Schoubroeck D, Timmerman D, Van Assche FA, Fryns JP. Associated malformations and chromosomal anomalies in 42 cases of prenatally diagnosed diaphragmatic hernia. Am J Med Genet. 2001;103:278–82. [PubMed: 11746006]
- Wright JC, Budd JL, Field DJ, Draper ES. Epidemiology and outcome of congenital diaphragmatic hernia: a 9-year experience. Paediatr Perinat Epidemiol. 2011;25:144–9. [PubMed: 21281327]
- Wung JT, Sahni R, Moffitt ST, Lipsitz E, Stolar CJ. Congenital diaphragmatic hernia: survival treated with very delayed surgery, spontaneous respiration, and no chest tube. J Pediatr Surg. 1995;30:406–9. [PubMed: 7760230]
- Yu L, Wynn J, Cheung YH, Shen Y, Mychaliska GB, Crombleholme TM, Azarow KS, Lim FY, Chung DH, Potoka D, Warner BW, Bucher B, Stolar C, Aspelund G, Arkovitz MS, Chung WK. Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic hernia. Hum Genet. 2013;132:285–92. [PMC free article: PMC3570587] [PubMed: 23138528]
- Zhang R, Thiele H, Bartmann P, Hilger AC, Berg C, Herberg U, Klingmüller D, Nürnberg P, Ludwig M, Reutter H. Whole-exome sequencing in nine monozygotic discordant twins. Twin Res Hum Genet. 2016;19:60–5. [PubMed: 26681452]
Publication Details
Author Information and Affiliations
Assistant, Massachusetts General Hospital
Boston, Massachusetts
Professor Emeritus of Pediatrics, Harvard Medical School
Boston, Massachusetts
MassGeneral Hospital for Children
Department of Surgery
Boston Children’s Hospital
Boston, Massachusetts
Publication History
Initial Posting: February 1, 2006; Last Revision: November 5, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Longoni M, Pober BR, High FA. Congenital Diaphragmatic Hernia Overview. 2006 Feb 1 [Updated 2020 Nov 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.