Summary
Clinical characteristics.
The term "galactosemia" refers to disorders of galactose metabolism that include classic galactosemia, clinical variant galactosemia, and biochemical variant galactosemia (not covered in this chapter). This GeneReview focuses on:
- Classic galactosemia, which can result in life-threatening complications including feeding problems, failure to thrive, hepatocellular damage, bleeding, and E coli sepsis in untreated infants. If a lactose-restricted diet is provided during the first ten days of life, the neonatal signs usually quickly resolve and the complications of liver failure, sepsis, and neonatal death are prevented; however, despite adequate treatment from an early age, children with classic galactosemia remain at increased risk for developmental delays, speech problems (termed childhood apraxia of speech and dysarthria), and abnormalities of motor function. Almost all females with classic galactosemia manifest hypergonadatropic hypogonadism or premature ovarian insufficiency (POI).
- Clinical variant galactosemia, which can result in life-threatening complications including feeding problems, failure to thrive, hepatocellular damage including cirrhosis, and bleeding in untreated infants. This is exemplified by the disease that occurs in African Americans and native Africans in South Africa. Persons with clinical variant galactosemia may be missed with newborn screening as the hypergalactosemia is not as marked as in classic galactosemia and breath testing is normal. If a lactose-restricted diet is provided during the first ten days of life, the severe acute neonatal complications are usually prevented. African Americans with clinical variant galactosemia and adequate early treatment do not appear to be at risk for long-term complications, including POI.
Diagnosis/testing.
The diagnosis of classic galactosemia and clinical variant galactosemia is established by detection of elevated erythrocyte galactose-1-phosphate concentration, reduced erythrocyte galactose-1-phosphate uridylyltranserase (GALT) enzyme activity, and/or biallelic pathogenic variants in GALT.
In classic galactosemia, erythrocyte galactose-1-phosphate is usually >10 mg/dL and erythrocyte GALT enzyme activity is absent or barely detectable. In clinical variant galactosemia, erythrocyte GALT enzyme activity is close to or above 1% of control values but probably never >10%-15%. However, in African Americans with clinical variant galactosemia, the erythrocyte GALT enzyme activity may be absent or barely detectable but is often much higher in liver and in intestinal tissue (e.g., 10% of control values).
Virtually 100% of infants with classic galactosemia or clinical variant galactosemia can be detected in newborn screening programs that include testing for galactosemia in their panel. However, infants with clinical variant galactosemia may be missed if the program only measures blood total galactose level and not erythrocyte GALT enzyme activity.
Management.
Treatment of manifestations: Standard of care in any newborn who is "screen-positive" for galactosemia is immediate dietary intervention while diagnostic testing is under way. Once a diagnosis is confirmed, restriction of galactose intake is continued and all milk products are replaced with lactose-free formulas (e.g., Isomil® or Prosobee®) containing non-galactose carbohydrates; dietary restrictions on all lactose-containing foods and other dairy products should continue throughout life, although management of the diet becomes less important after infancy and early childhood. In rare instances, cataract surgery may be needed in the first year of life. Childhood apraxia of speech and dysarthria require expert speech therapy. Developmental assessment at age one year by a psychologist and/or developmental pediatrician is recommended in order to formulate a treatment plan with the speech therapist and treating physician. For school-age children, an individual education plan and/or professional help with learning skills and special classrooms as needed. Hormone replacement therapy as needed for delayed pubertal development and/or primary or secondary amenorrhea. Stimulation with follicle-stimulating hormone may be useful in producing ovulation in some women.
Prevention of secondary complications: Recommended calcium, vitamin D, and vitamin K intake to help prevent decreased bone mineralization; standard treatment for gastrointestinal dysfunction.
Surveillance: Biochemical genetics clinic visits every three months for the first year of life or as needed depending on the nature of the potential acute complications; every six months during the second year of life; yearly thereafter. Routine monitoring for: the accumulation of toxic analytes (e.g., erythrocyte galactose-1-phosphate and urinary galactitol); cataracts; speech and development; movement disorder; POI; nutritional deficiency; and osteoporosis.
Agents/circumstances to avoid: Breast milk, proprietary infant formulas containing lactose, cow's milk, dairy products, and casein or whey-containing foods; medications with lactose and galactose.
Evaluation of relatives at risk: To allow for earliest possible diagnosis and treatment of at-risk sibs:
- Perform prenatal diagnosis when the GALT pathogenic variants in the family are known; or
- If prenatal testing has not been performed, test the newborn for either the family-specific GALT pathogenic variants or erythrocyte GALT enzyme activity.
Pregnancy management: Women with classic galactosemia should maintain a lactose-restricted diet during pregnancy.
Genetic counseling.
Classic galactosemia and clinical variant galactosemia are inherited in an autosomal recessive manner. Couples who have had one affected child have a 25% chance of having an affected child in each subsequent pregnancy. Molecular genetic carrier testing for at-risk sibs and prenatal testing for pregnancies at increased risk are an option if the GALT pathogenic variants in the family are known. If the GALT pathogenic variants in a family are not known, prenatal testing can rely on assay of GALT enzyme activity in cultured amniotic fluid cells.
GeneReview Scope
Table
Classic galactosemia Clinical variant galactosemia
Diagnosis
An international clinical guideline addressing management has been published [Welling et al 2017] (full text).
Scenarios
Scenario 1 – Abnormal Newborn Screening (NBS) Result
NBS for classic galactosemia and clinical variant galactosemia is primarily based on quantification of the following analytes and/or enzymatic testing, depending on the state:
- Total content of erythrocyte galactose-1-phosphate and blood galactose concentration; and/or
- Erythrocyte GALT enzyme activity
Total erythrocyte galactose-1-phosphate and blood galactose concentration values above the cutoff reported by the screening laboratory are considered positive and require follow-up testing in a biochemical genetics laboratory.
In classic galactosemia
- Erythrocyte galactose-1-phosphate may be as high as 120 mg/dL, but is usually >10 mg/dL in the newborn period. When the affected individual is on a lactose-free diet, the level is ≥1.0 mg/dL. Normal level of erythrocyte galactose-1-phosphate is <1 mg/dL.
- Plasma free galactose is usually >10 mg/dL, but may be as high as 90-360 mg/dL (5-20 mmol/L).
- Galactose-1-phosphate uridylyltranserase (GALT) enzyme activity is absent or barely detectable.
In clinical variant galactosemia
- Erythrocyte galactose-1-phosphate is usually >10 mg/dL. When the affected individual is on a lactose-free diet, the level is usually <1.0 mg/dL. Normal level of erythrocyte galactose-1-phosphate is <1 mg/dL.
- Plasma free galactose is usually >10 mg/dL, but may be as high as 90-360 mg/dL (5-20 mmol/L).
- Erythrocyte GALT enzyme activity is close to or above 1% of control values but probably never >10%-15%.Note: In African Americans with clinical variant galactosemia, the erythrocyte GALT enzyme activity may be absent or barely detectable but is often much higher in liver and in intestinal tissue (e.g., 10% of control values).In certain populations (e.g., African Americans with hypomorphic alleles including p.Ser135Leu/Ser135Leu), erythrocyte GALT enzyme activity may be absent or barely detectable.
Dietary intervention – including replacing all milk products with lactose-free formulas (e.g., Isomil® or Prosobee®) containing non-galactose carbohydrates – needs to begin immediately on receipt of an abnormal NBS result while additional testing is performed to determine whether it is a true positive NBS result and to establish the diagnosis of classic galatosemia or clinical variant galatosemia definitively.
Scenario 2 – Symptomatic Individual
A symptomatic individual who has either atypical findings, untreated infantile-onset classic galactosemia, or clinical variant galactosemia may present as a result of any of the following: NBS not performed; false negative NBS result; caregivers not adherent to recommended treatment following a positive NBS result.
Supportive clinical findings can include the following:
- Untreated infant:
- Feeding problems
- Failure to thrive
- Liver failure
- Bleeding
- E coli sepsis
- Untreated older person:
- Developmental delay
- Speech problems
- Abnormalities of motor function, including extrapyramidal findings with ataxia
- Cataracts
- Liver failure/cirrhosis
- Premature ovarian failure in females
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of classic galactosemia or clinical variant galactosemia is established in a proband by detection of elevated erythrocyte galactose-1-phosphate concentration, reduced erythrocyte galactose-1-phosphate uridylyltranserase (GALT) enzyme activity, and/or biallelic pathogenic variants in GALT (Table 1).
Molecular genetic testing approaches include single-gene testing:
- Sequence analysis of GALT is performed first and followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
- Targeted analysis for common pathogenic variants can be performed first in individuals of European or African ancestry (see Table 1). This approach is most efficient when testing large numbers of samples (e.g., carrier screening or newborn screening).
Table 1.
Molecular Genetic Testing Used in Classic Galactosemia and Clinical Variant Galactosemia
Clinical Characteristics
Clinical Description
Galactosemia caused by deficiency of the enzyme galactose-1-phosphate uridylyltranserase (GALT) may be divided into three clinical/biochemical phenotypes: (1) classic galactosemia, (2) clinical variant galactosemia, and (3) biochemical variant galactosemia (not covered in this GeneReview; see, for example, Duarte Variant Galactosemia). This categorization is based on residual erythrocyte GALT enzyme activity; the levels of galactose metabolites (e.g., erythrocyte galactose-1-phosphate and urine galactitol) that are observed both off and on a lactose-restricted diet; and, most importantly, the likelihood that the affected individual will develop acute and chronic long-term complications. This categorization allows for proper counseling of the parents of an infant with galactosemia, especially regarding the so-called diet-independent complications.
Classic Galactosemia
Scenario 1 – Abnormal newborn screening (NBS) result and prompt initiation of appropriate management in the neonatal period
Within days of ingesting breast milk or lactose-containing formulas, infants with classic galactosemia develop life-threatening complications including feeding problems, failure to thrive, hypoglycemia, hepatocellular damage, bleeding diathesis, and jaundice (see Table 2).
If a lactose-free diet is provided during the first three to ten days of life, the signs of classic galactosemia resolve quickly and prognosis for prevention of liver failure, E coli sepsis, and neonatal death is good. Failure to implement effective newborn screening may have catastrophic consequences such as liver failure [Malone et al 2011].
Long-term outcome. Even with early and adequate therapy, the long-term outcome in older children and adults with classic galactosemia can include cataracts, speech defects, poor growth, poor intellectual function, neurologic deficits (predominantly extrapyramidal findings with ataxia), and, in females, hypergonadotropic hypogonadism or premature ovarian insufficiency (POI) [Rubio-Gozalbo et al 2019].
Classic galactosemia is associated with extreme variability in chronic complications and/or long-term outcome. Even individuals who have not been sick in the newborn period and who were begun on a lactose-free diet from birth (e.g., those with a prior affected sib in the family) may manifest language delay, speech defects, learning disabilities, cognitive impairment, and in females, POI. These problems may manifest as early as age one to two years, and in almost all instances, no findings that would have predicted eventual brain and ovarian dysfunction were present in early infancy. A minority of individuals may exhibit documented neurologic abnormalities including tremor (postural or intentional), cerebellar ataxia, and dystonia. No findings early in the disease course are good predictors of these long-term complications. Overall, the quality of life is reduced in adults with classic galactosemia, and more so when compared to individuals with phenylketonuria (PKU) [Gubbels et al 2011, ten Hoedt et al 2011, Hoffmann et al 2012].
Scenario 2 – Symptomatic individual with untreated classic galactosemia (resulting from NBS not performed or false negative NBS result)
If classic galactosemia is not treated, sepsis with E coli, shock, and death may occur [Levy et al 1977]. Infants who survive the neonatal period and continue to ingest lactose may develop severe brain damage [Otaduy et al 2006].
Table 2.
Frequency of Specific Findings in Symptomatic Neonates with Classic Galactosemia
If the diagnosis of classic galactosemia is not established, the infant who is partially treated with intravenous antibiotics and self-restricted lactose intake demonstrates relapsing and episodic jaundice and bleeding from altered hemostasis concomitant with the introduction of lactose. If treatment is delayed, complications such as growth restriction and progressive liver disease are likely. Rare affected individuals may develop vitreous hemorrhages that may produce blindness [Levy et al 1996, Takci et al 2012].
Long-term outcome. Information on long-term outcomes was initially reported by Waggoner et al [1990] as the result of a retrospective, cross-sectional survey of 270 individuals with classic galactosemia. The largest outcome study involving 507 people with classic galactosemia was published by the international GalNet registry or consortium [Rubio-Gozalbo et al 2019]. To summarize, the data on long-term outcome indicate that complications involving the nervous system and ovarian function do not correlate with any of the well-known biochemical variables (e.g., erythrocyte galactose-1-phosphate levels); furthermore, manifestations of one or more of these complications vary even among individuals with the same genotype associated with classic galactosemia (see Table 3) [Doyle et al 2010, Schadewaldt et al 2010, Hoffmann et al 2011, Krabbi et al 2011, Coss et al 2012, Waisbren et al 2012, Frederick et al 2017].
- Intellectual development. Of 177 individuals age six years or older with no obvious medical causes for developmental delay other than galactosemia, 45% were described as developmentally delayed.
- The mean IQ scores of the individuals as a group declined slightly (4-7 points) with increasing age.
- Studies of Dutch individuals at various ages using a quality-of-life questionnaire indicated subnormal cognitive outcomes [Bosch et al 2004b].
- Speech problems were reported in 56% (136/243) of individuals age three years or older.
- More than 90% of individuals with speech problems were described as having delayed vocabulary and articulation problems. The speech problems resolved in only 24%.
- A more formal analysis found speech problems in 44% of individuals; 38% had a specific diagnosis including childhood apraxia of speech [Robertson & Singh 2000, Webb et al 2003].
- Speech defects are heterogeneous (involving both central defects and motor abnormalities) and evolve with time [Potter et al 2013].
- The developmental quotients and IQ scores observed in individuals with speech disorders as a group were significantly lower than those of individuals with normal speech, although some individuals with speech problems tested in the average range.
- Motor function. Among individuals older than age five years, 18% had fine motor tremors and problems with coordination, gait, and balance.
- Severe ataxia was observed in two teenagers.
- Adults manifested tremors, dysarthria, cerebellar ataxia, and dystonia [Waisbren et al 2012, Rubio-Agusti et al 2013].
- Gonadal function in females. Of 47 girls and women, 81% had signs of POI.
- The mean age at menarche was 14 years with a range from ten to 18 years.
- Eight of 34 women older than age 17 years (including 2 with "streak gonads") had primary amenorrhea.
- Most women developed oligomenorrhea and secondary amenorrhea within a few years of menarche.
- Only five of 17 women older than age 22 years had normal menstruation. Two, who gave birth at ages 18 and 26 years, had never experienced normal menstrual periods.
- Guerrero et al [2000] determined that the development of POI in females with galactosemia is more likely if the following are true:
- The individual is homozygous for p.Gln188Arg.
- The mean erythrocyte galactose-1-phosphate concentration is >3.5 mg/dL during therapy.
- The recovery of 13CO2 from whole-body 13C galactose oxidation is reduced below 5% of administered 13C galactose.
- Gonadal function in males
- Normal serum concentrations of testosterone and/or follicle-stimulating hormone and luteinizing hormone were reported for males. However, the literature has few reports of males with classic galactosemia who have fathered a child [Panis et al 2006a, Waisbren et al 2012, Gubbels et al 2013].
- There have been no data to support structural abnormalities in the male reproductive tract that would lead to infertility; preliminary data suggest an increased prevalence of cryptorchidism and low semen volume [Gubbels et al 2013].
- Growth. In many individuals, growth was severely delayed during childhood and early adolescence; when puberty was delayed and growth continued through the late teens, final adult heights were within the normal range. Decreased height over mean parental height was related to low insulin-like growth factor-I [Panis et al 2007].
- Cataracts were reported in 30% of 314 individuals.
- Nearly half the cataracts were described as "mild," "transient," or "neonatal" and resolved with dietary treatment; only eight were treated surgically.
- Dietary treatment had begun at a mean age of 77 days for those with cataracts compared to 20 days for those without cataracts. However, one of the eight individuals who required cataract surgery was an infant who had been treated from birth.
Relationship between treatment and outcome. No significant associations were found between treatment and outcome except for a greater incidence of developmental delay among individuals who were not treated until after age two months. However, IQ scores were not highly correlated with the age at which treatment began. The effect of early treatment on outcome was also studied in 27 sibships, three of which had three affected sibs. The older sibs were diagnosed and treated after clinical symptoms occurred or newborn screening results had been reported, whereas the younger sibs were treated within two days of birth. Although the younger sibs were treated early and only one developed neonatal symptoms, the differences in IQ scores among the sibs were not statistically significant, and the speech and ovarian function of the younger sibs were no better than those of their older sibs.
- Restriction of milk in the mother's diet during pregnancy was reported for 21 of the 38 infants who were treated from birth. The long-term outcome of these 21 was no better than that of the 17 individuals whose intake of mother's milk was not restricted during the pregnancy.
- No significant differences could be observed in the rate of complications between the individuals with residual enzyme activity and those with no measurable enzyme activity, except that individuals with some enzyme activity tended to be taller for age.
Individuals with or without neurologic complications. No differences were observed in treatment or biochemical factors between the 56 individuals with normal intellect, speech, and motor function and the 25 individuals with developmental delay and speech and motor problems.
Relationships of complications. Developmental delay and low IQ scores were associated with speech problems, motor problems, and delayed growth, but not with abnormal ovarian function.
Sex differences. Females had lower mean IQ scores than males after age ten years (p<0.05) and had lower mean heights for age at five to 12 years (p<0.05), but did not differ in frequency of speech or motor problems or in the treatment variables, including age at which treatment began, neonatal illness, or galactose-1-phosphate erythrocyte concentration. However, the association of problems with intellectual development, speech, and motor function could also indicate a specific neurologic abnormality in some cases of galactosemia [Schadewaldt et al 2010].
Clinical Variant Galactosemia
Individuals with variant forms of galactosemia may have some aspects of classic galactosemia, including early cataracts, liver disease, mild intellectual disability with ataxia, and growth restriction [Fridovich-Keil et al 2011]. Clinical variant galactosemia can result in life-threatening complications in untreated infants, including feeding problems, failure to thrive, hepatocellular damage (including cirrhosis), and bleeding.
Clinical variant galactosemia is exemplified by the disease that occurs in African Americans and native Africans in South Africa with a p.Ser135Leu/Ser135Leu genotype. Neonates with clinical variant galactosemia may be missed with NBS because the hypergalactosemia is not as marked as in classic galactosemia and breath testing is normal [Crushell et al 2009].
If a lactose-restricted diet is provided during the first ten days of life, the severe acute neonatal complications usually do not occur.
To the best of current knowledge, African Americans with clinical variant galactosemia and adequate early treatment do not develop long-term complications, including POI.
Genotype-Phenotype Correlations
Significant genotype-phenotype correlations have been noted [Shield et al 2000, Tyfield 2000, Demirbas et al 2019]. Although the GALT genotype informs prognosis [Guerrero et al 2000, Webb et al 2003], some confusion about genotype-phenotype correlations appears to have resulted from the variability of the manifestations and severity of the chronic complications of classic galactosemia.
Use of the galactosemia classification system in Table 3, employed in the study by Demirbas et al [2019], helps dispel confusion. The most common pathogenic variants that result in the three galactosemia phenotypes – classic, clinical variant, and biochemical variant – are shown in Table 3. (See Diagnosis and Genetically Related Disorders for definitions.)
Table 3.
GALT Genotypes and Biochemical/Clinical Phenotypes
p.Gln188Arg. Approximately 70% of the alleles in persons with GALT deficiency of northern European ancestry have a substitution of an arginine for a glutamine at amino acid position 188.
- In the homozygous state, the pathogenic variant interferes with the catalytic reaction. It is associated with increased risks for POI and childhood apraxia of speech [Robertson & Singh 2000].
- In one cross-sectional retrospective study correlating genotype with outcome in individuals with classic galactosemia, a greater proportion of individuals with a poor outcome were homozygous for the p.Gln188Arg pathogenic variant, and a greater proportion with a good outcome were not homozygous for the p.Gln188Arg pathogenic variant. However, one adult female and one adult male homozygous for the p.Gln188Arg pathogenic variant who had begun normal lactose intake at age three years exhibited no worsening of the underlying classic galactosemia phenotype [Lee et al 2003, Panis et al 2006a].
p.Ser135Leu, in which a leucine is substituted for serine at amino acid 135, is prevalent in Africa.
- If therapy is initiated in the neonatal period, African Americans with galactosemia who have this allele in the homozygous state have a good prognosis. Generally, these individuals are not prone to E coli sepsis in the neonatal period or chronic complications (i.e., speech disorder, POI, and intellectual disability) when treated from infancy [Lai et al 1996].
- Data are limited on outcome in persons who are compound heterozygous (p.[Ser135Leu];[Gln188Arg]); however, they appear to have fewer complications than individuals with the genotype p.[Gln188Arg]+[p.Gln188Arg], associated with classic galactosemia.
p.Asn314Asp. The Duarte (D2) variant is the allele in which an aspartate is substituted for asparagine at residue 314 and a second variant in cis configuration (a 4-bp deletion in the promoter region: c.-119_116delGTCA) results in reduced erythrocyte GALT enzyme activity. The D2 allele designation is c.[940A>G;-119_116delGTCA].
- In the homozygous state D2 erythrocyte GALT enzyme activity is reduced by 50%.
- Compound heterozygotes with D2 and a pathogenic variant associated with classic galactosemia have a good prognosis [Langley et al 1997, Lai et al 1998].
- However, compound heterozygosity for D2 (Duarte) and D1 (LA variant) is known to occur. The D1 allele has a p.Leu218Leu in cis configuration with the p.Asn314Asp pathogenic variant p.[Leu218Leu;Asn314Asp], which confers "superactivity" (i.e., heterozygotes have ~117% erythrocyte GALT activity while homozygotes display ~134% activity).
Other. Substitution of an asparagine for a lysine at position 285 (p.Lys285Asn) is prevalent in southern Germany, Austria, and Croatia; it is associated with a poor prognosis for neurologic and cognitive function in either the homozygous state or compound heterozygous state with p.Gln188Arg and is considered classic galactosemia.
Other compound heterozygotes (e.g., p.[Gln188Arg]+[Arg333Gly]) have a good long-term outcome [Ng et al 2003].
- A clear genotype-phenotype correlation is seen when classic galactosemia with genotypes such as p.[Gln188Arg]+[Gln188Arg] is compared with clinical variant galactosemia caused by the p.[Ser135Leu]+[Ser135Leu] genotype. For example, almost all females with the p.[Gln188Arg]+[Gln188Arg] genotype manifest POI, whereas POI is almost unheard of in African American women with the p.[Ser135Leu]+[Ser135Leu] genotype. A critical unanswered question is how much residual GALT enzyme activity in target tissues there must be to eliminate chronic diet-independent complications. To illustrate, cryptic residual GALT enzyme activity may be a potential modifier of scholastic outcome in school-age children [Ryan et al 2013].
- While on a lactose-restricted diet, persons with classic galactosemia display erythrocyte galactose-1-phosphate levels between 1 and 5 mg/dL and urine galactitol levels between 100 and 400 μmol/mmol creatinine, whereas persons with a p.[Ser135Leu]+[Ser135Leu] genotype usually have an erythrocyte galactose-1-phosphate level below 1 mg/dL, and urine galactitol that is below 100 μmol/mmol creatinine and often in the normal range [Saudubray et al 2012, Walter & Fridovich-Keil 2014].
- Persons with biochemical variant galactosemia – for example, compound heterozygotes for c.563A>G (p.Gln188Arg) and D2 c.[940A>G;-119_116delGTCA] genotype – differ from those with either classic galactosemia or clinical variant galactosemia: they generally exhibit no signs and symptoms of disease, only biochemical perturbations.
Note: Many of the more than 300 GALT pathogenic variants have been identified following newborn screening with little or no long-term follow-up data. In these instances, the term "classic galactosemia" should be applied with caution or not at all. It would be inappropriate to counsel new parents that their infant will develop one or more of the chronic complications seen in galactosemia without supportive data.
Nomenclature
Table 4.
The Genetic Hypergalactosemias
Prevalence
Based on the results of newborn screening programs, the prevalence of classic galactosemia is 1:48,000 [National Newborn Screening and Genetics Resource Center 2014]. However, when erythrocyte GALT enzyme activity <5% of control activity and erythrocyte galactose-1-phosphate concentration >2 mg/dL are used as diagnostic criteria, some newborn screening programs record a prevalence of 1:10,000 [Bosch et al 2005].
The frequency of classic galactosemia in Ireland is 1:16,476 [Coss et al 2013].
While it is not possible to provide prevalence data for clinical variant galactosemia, the estimated prevalence of the p.Ser135Leu/Ser135Leu genotype is 1:20,000 [Henderson et al 2002].
Genetically Related (Allelic) Disorders
Duarte variant galactosemia, an example of biochemical variant galactosemia, is associated with specific pathogenic variants in GALT.
The Duarte variant (D2) has in cis configuration (i.e., in the same allele) the pathogenic missense variant p.Asn314Asp and a GTCA deletion in the promoter region (c.-119-116delGTCA) that impairs a positive regulatory domain. It is designated c.[940A>G;-119_116delGTCA] (see Table 3).
Note: The Los Angeles (LA) variant (D1) has the identical p.Asn314Asp pathogenic missense variant as the Duarte variant but does not have the GTCA promoter deletion. Instead, it is in cis configuration with the missense variant p.Leu218Leu. This variant does not cause galactosemia and is associated with increased erythrocyte GALT enzyme activity [Langley et al 1997, Elsas et al 2002].
In biochemical variant galactosemia:
- Erythrocyte galactose-1-phosphate is usually >1 mg/dL, but may be as high as 35 mg/dL. When the individual is on a lactose-free diet, the level is <1 mg/dL.
- Residual erythrocyte GALT enzyme activity is usually >15% and, on average, is 25% of control values.
Duarte variant galactosemia, associated with one D2 c.[940A>G;-119_116delGTCA] and one severe pathogenic variant, is the most prevalent form of biochemical variant galactosemia. While individuals with this condition may display elevated galactose levels, most experts agree that dietary therapy is not required [Carlock et al 2019].
Differential Diagnosis
The differential diagnosis for neonatal hepatotoxicity includes infectious diseases, obstructive biliary disease, and metabolic diseases; see Table 4 for selected examples. See Table 5 for other genetic hypergalactosemias.
Note: Establishing the diagnosis of sepsis does not exclude the possibility of galactosemia, as sepsis (particularly E coli sepsis) occurs commonly in infants with classic galactosemia.
Table 5.
Genes of Interest in the Differential Diagnosis of Neonatal Hepatotoxicity
Table 6.
Genes of Interest in the Differential Diagnosis of Hypergalactosemias
Management
When classic galactosemia or clinical variant galactosemia is suspected during the diagnostic evaluation (i.e., due to a positive newborn screen or based on clinical features), dietary intervention should be initiated immediately. An international clinical guideline addressing management has been published [Welling et al 2017] (full text).
Evaluations Following Initial Diagnosis in the Newborn Period
To establish the extent of disease and needs in an individual diagnosed with classic galactosemia or clinical variant galactosemia, the evaluations summarized in Table 7 are recommended.
Table 7.
Recommended Evaluations Following Initial Diagnosis of Classic Galactosemia and Clinical Variant Galactosemia
Treatment of Manifestations and Complications
An international clinical guideline addressing management has been published [Welling et al 2017] (full text).
Table 8.
Treatment in Individuals with Classic Galactosemia and Clinical Variant Galactosemia
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech therapy services will be provided within the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction. Physical therapy is recommended to maximize mobility.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Communication issues. Affected individuals with childhood apraxia of speech and dysarthria require therapy by a speech expert until the complication has been brought under control. This may require years of intensive therapy.
Prevention of Primary Manifestations
See Table 8.
Prevention of Secondary Complications
Because bone mineral density in children and adults with classic galactosemia and clinical variant galactosemia may be diminished, supplements of vitamin D in excess of 1,000 IU/day and vitamin K have been recommended [Panis et al 2006b, Batey et al 2013].
- For calcium and vitamin D intake recommendations, see the NIH Osteoporosis and Related Bone Diseases website.
- If routine follow-up visits with a dietitian knowledgeable about metabolic disorders have verified that calcium and vitamin D intake are adequate for age and if plasma 25-hydroxyvitamin D is within the normal range but bone mineral density is decreased, consultation with a pediatric and/or adult endocrinologist may be warranted.
Some affected individuals may develop gastrointestinal dysfunction such as chronic constipation. For those with severe symptoms, evaluation by a gastroenterologist may be considered [Shaw et al 2017].
Surveillance
See also the management guidelines published by Welling et al [2017] (full text).
Schedule for Individuals with Classic or Clinical Variant Galactosemia
Biochemical genetics clinic visits
- Every three months for the first year of life or as needed depending on the nature of potential acute complications
- Every six months during the second year of life
- Yearly thereafter
Metabolic dietician clinic visits are indicated as above, plus interim visits and phone consultations as necessary.
In addition to regular evaluations by a metabolic specialist and metabolic dietician, the evaluations in Table 9 are recommended.
Table 9.
Recommended Surveillance for Individuals with Classic Galactosemia and Clinical Variant Galactosemia
Agents/Circumstances to Avoid
The following should be avoided:
- Breast milk, proprietary infant formulas containing lactose, cow's milk, dairy products, and casein- or whey-containing foods
- Lactose- or galactose-containing drug preparations
- Medicines that contain lactose (tablets, capsules, sweetened elixirs), especially during infancy
Evaluation of Relatives at Risk
Prenatal testing of a fetus at risk. When the pathogenic variants causing classic galactosemia or clinical variant galactosemia in the family are known, prenatal testing of fetuses at risk may be performed via amniocentesis or chorionic villus sampling to allow for institution of treatment at birth.
Newborn sib. If prenatal testing has not been performed, each at-risk newborn sib should be treated from birth and screened for classic galactosemia or clinical variant galactosemia using the erythrocyte GALT enzyme assay and/or genetic testing (if the familial pathogenic variants in the family are known) to allow for earliest possible diagnosis. Note: If there are concerns about the reliability of the prenatal testing, soy-based formula may be given while the diagnostic testing is being performed.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Affected females. Women with classic galactosemia or clinical variant galactosemia should be on a lactose-restricted diet during pregnancy.
Carrier females. There is no evidence that the outcome of children with classic galactosemia or clinical variant galactosemia is improved if their mothers (who are obligate carriers) were on a lactose-restricted diet during pregnancy. Therefore, unaffected pregnant women who are heterozygous for a pathogenic variant in GALT (carrier females) do NOT require a lactose-restricted diet during pregnancy.
Therapies Under Investigation
Research suggests that despite exogenous galactose restriction, endogenous galactose production may approach 1-2 g/day [Berry et al 2004, Schadewaldt et al 2004]. If this is true, "self-intoxication" with galactose may be more of a problem than restriction of galactose from exogenous sources in the management of older children and adults who no longer depend on milk as their primary source of energy.
Approaches to lowering endogenous production of galactose-1-phosphate are under investigation using small inhibitors of the GALK enzyme [Tang et al 2010]. While in vitro studies of GALT enzyme-deficient human fibroblasts demonstrated proof of concept, it is yet to be performed in an animal model. The GALT knockout mice generated by Leslie et al [1992] do not express the human phenotype of galactosemia and – except for polyuria due to hypergalactosemia and hypergalactosuria – are largely without disease. As mice have lost ARHI (DIRAS3) during evolution [Lai et al 2008, Rubio-Gozalbo et al 2010, Tang et al 2010], a GALT enzyme-deficient mouse model that expresses an ARHI (DIRAS3) signal is needed to test the hypothesis that ARHI gene expression plays a role in phenotypic expression of disease and to determine whether inhibition of galactose-1-phosphate production limits or abrogates the "human phenotype."
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Classic galactosemia and clinical variant galactosemia are inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are obligate heterozygotes (i.e., presumed to be carriers of one GALT pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a GALT pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and do not develop galactosemia.
Sibs of a proband
- If both parents are known to be heterozygous for a GALT pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and do not develop galactosemia.
Offspring of a proband
- The offspring of an individual with classic galactosemia or clinical variant galactosemia are obligate heterozygotes (carriers) for a pathogenic variant in GALT.
- If one parent has classic galactosemia or clinical variant galactosemia and the other parent is a carrier, each child has a 50% chance of being a heterozygote and a 50% chance of having classic galactosemia or clinical variant galactosemia.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a GALT pathogenic variant.
Carrier Detection
Molecular genetic testing. Carrier testing for at-risk relatives requires prior identification of the GALT pathogenic variants in the family.
Biochemical genetic testing. Carrier testing is done by measuring erythrocyte GALT enzyme activity. Erythrocyte GALT enzyme activity is:
- ~50% of control values in carriers of classic galactosemia;
- ~50% of control values in carriers of p.Ser135Leu-related clinical variant galactosemia; this will be different with other clinical variant galactosemia-causing pathogenic variants, which in the homozygous state result in 1%-10% residual erythrocyte GALT enzyme activity.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once both GALT pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for classic galactosemia or clinical variant galactosemia are possible.
Prenatal testing (or preimplantation genetic testing) using molecular genetic testing is preferred over enzyme analysis.
Biochemical testing. Analysis of GALT enzyme activity and molecular diagnosis rely on cells obtained by chorionic villus sampling or amniocentesis.
Note: When a fetus has classic galactosemia or clinical variant galactosemia, amniotic fluid concentration of galactitol is elevated in the late third trimester and has been used in the past for prenatal testing.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- British Inherited Metabolic Disease Group (BIMDG)TEMPLE (Tools Enabling Metabolic Parents LEarning)United Kingdom
- Galactosemia Support Group (GSG)31 Cotysmore RoadSutton Coldfield West Midlands B75 6BJUnited KingdomPhone: 0121 378 5143Email: contact@galactosaemia.org
- Medical Home Portal
- National Library of Medicine Genetics Home Reference
- The Galactosemia Foundation350 Northern BoulevardSuite 324 - 1079Albany NY 12204-1000Phone: 866-900-7421Email: outreach@galactosemia.org
- Apraxia KidsPhone: 412-785-7072Email: info@apraxia-kids.org
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Newborn Screening in Your StateHealth Resources & Services Administration
For information about the GalNet Registry, see Chapter Notes.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Classic Galactosemia and Clinical Variant Galactosemia: Genes and Databases
Table B.
OMIM Entries for Classic Galactosemia and Clinical Variant Galactosemia (View All in OMIM)
Molecular Pathogenesis
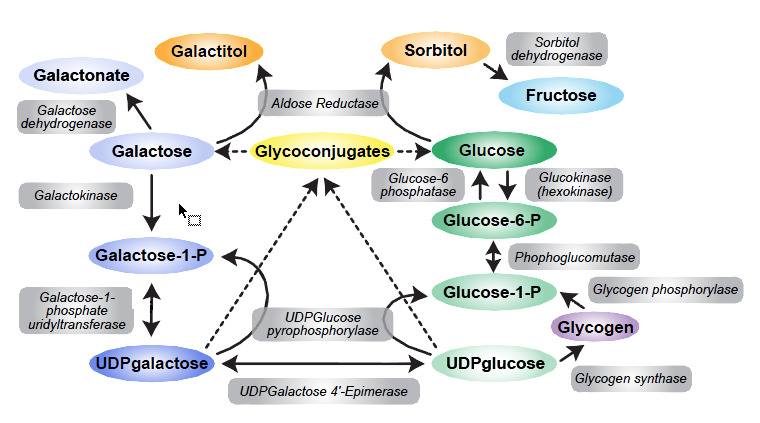
The GALT enzyme catalyzes the conversion of galactose-1-phosphate and UDPglucose to UDPgalactose and Glu-1-P in a two-step process termed a ping-pong or bi-bi molecular reaction (Figure 1):
Figure 1.
Galactose metabolism, the Leloir pathway
- 1.
UDPglucose binds to the active site and glucose-1-phosphate is released, leaving UMP covalently linked to the enzyme.
- 2.
Galactose-1-phosphate then lands at the active site, engages the bound UMP, and following cleavage of the phosphonium bond, UDP galactose is released.
When GALT enzyme activity is deficient, galactose-1-phosphate, galactose, and galactitol accumulate (Figure 2). Galactose is converted to galactitol in cells and produces osmotic effects including swelling of lens fibers that may result in cataracts. The same process has been hypothesized to produce swelling of brain cells and subsequently, pseudotumor cerebri.
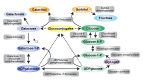
Figure 2.
Galactose metabolism, GALT deficiency
Since individuals with classic galactosemia who are prospectively treated may manifest all of the so-called chronic diet-independent complications, and since the amniotic fluid of affected fetuses contains high levels of galactitol and the cord blood of affected newborns contains elevated levels of erythrocyte galactose-1-phosphate, one must consider whether the long-term complications of GALT enzyme deficiency are due to prenatal toxicity [Komrower 1982]. One hypothesis is that the prenatal central nervous system insult is secondary to myo-inositol deficiency [Berry 2011].
Mechanism of disease causation. Loss of function
Table 10.
Notable GALT Variants
Chapter Notes
The GalNet Registry is maintained as a research tool by a steering committee composed of investigators from the USA and Europe. It is not currently open to the public.
Author Notes
Gerard T Berry is the Harvey Levy Chair in Metabolism at the Boston Children's Hospital and Professor of Pediatrics at the Harvard Medical School. He is the Director of Metabolism Program at Boston Children's Hospital and the Director of the Harvard Medical School Biochemical Genetics Training Program. He is a member of the Broad Institute of Harvard and MIT. He was the recipient of the 2004 Emmanuel Shapiro Society for Inherited Metabolic Disorders (SIMD) Award. As a member of the SIMD Board of Directors, Dr Berry is the SIMD President Elect. He is the co-chair of the Undiagnosed Disease Network (UDN) metabolomics working group. Dr Berry has established an international center for galactosemia at Boston Children's Hospital. His research has been in both the clinical and basic science spheres. The Berry laboratory has been heavily involved in stable isotope breath testing and whole-body galactose modeling in patients with galactosemia. He created new accurate methods to quantify GALT, GALK, and GALE enzyme activities using LC-MS/MS methodology. More recently his laboratory has turned to studying human induced pluripotent stem (IPS) cells generated from blood and fibroblasts of individuals with galactosemia. He is using synthetic cortical neurons derived from IPS cells, as well as human organoids, to study the mechanisms of central nervous system dysfunction and cell death in galactosemia.
Acknowledgments
I thank the Galactosemia Foundation for their many years of support of this work, all of my colleagues and co-investigators, and the patients and families who have contributed so much and participated in the various research studies.
Author History
Gerard T Berry, MD, FFACMG (2014-present)
Louis J Elsas II, MD, FFACMG; University of Miami (1999-2014)
Revision History
- 11 March 2021 (ma) Comprehensive update posted live
- 9 March 2017 (ma) Comprehensive update posted live
- 3 April 2014 (me) Comprehensive update posted live
- 26 October 2010 (me) Comprehensive update posted live
- 27 September 2007 (me) Comprehensive update posted live
- 2 May 2005 (me) Comprehensive update posted live
- 27 March 2003 (me) Comprehensive update posted live
- 4 February 2000 (me) Review posted live
- 31 August 1999 (le) Original submission
References
Published Guidelines / Consensus Statements
- Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, Grünewald S, Gubbels CS, Knerr I, Labrune P, van der Lee JH, MacDonald A, Murphy E, Portnoi PA, Õunap K, Potter NL, Rubio-Gozalbo ME, Spencer JB, Timmers I, Treacy EP, Van Calcar SC, Waisbren SE, Bosch AM; Galactosemia Network (GalNet). International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. Available online. 2017. Accessed 3-24-22.
Literature Cited
- Barbouth D, Slepak T, Klapper H, Lai K, Elsas LJ (2006) Prevention of a molecular misdiagnosis in galactosemia. Genet Med 8:178-82 [PubMed: 16540753]
- Batey LA, Welt CK, Rohr F, Wessel A, Anastasoaie V, Feldman HA, Guo CY, Rubio-Gozalbo E, Berry G, Gordon CM. Skeletal health in adult patients with classic galactosemia. Osteoporos Int. 2013;24:501-9. [PubMed: 22525982]
- Berry GT. Galactosemia: when is it a newborn screening emergency? Mol Genet Metab. 2012;106:7-11. [PubMed: 22483615]
- Berry GT. Is prenatal myo-inositol deficiency a mechanism of CNS injury in galactosemia? J Inherit Metab Dis.2011;34:345-55. [PubMed: 21246399]
- Berry GT, Moate PJ, Reynolds RA, Yager CT, Ning C, Boston RC, Segal S (2004) The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab 81:22-30 [PubMed: 14728988]
- Bosch AM, Bakker HD, Wenniger-Prick LJ, Wanders RJ, Wijburg FA (2004a) High tolerance for oral galactose in classical galactosaemia: dietary implications. Arch Dis Child 89:1034-6 [PMC free article: PMC1719730] [PubMed: 15499058]
- Bosch AM, Grootenhuis MA, Bakker HD, Heijmans HS, Wijburg FA, Last BF (2004b) Living with classical galactosemia: health-related quality of life consequences. Pediatrics 113:e423-8 [PubMed: 15121984]
- Bosch AM, Ijlst L, Oostheim W, Mulders J, Bakker HD, Wijburg FA, Wanders RJ, Waterham HR (2005) Identification of novel mutations in classical galactosemia. Hum Mutat 25:502 [PubMed: 15841485]
- Carlock G, Fischer ST, Lynch ME, Potter NL, Coles CD, Epstein MP, Mulle JG, Kable JA, Barrett C, Edwards SM, Wilson E, Fridovich-Keil JL (2019) Developmental outcomes in Duarte galactosemia. Pediatrics. 143:e20182516 [PubMed: 30593450]
- Coffee B, Hjelm LN, DeLorenzo A, Courtney EM, Yu C, Muralidharan K (2006) Characterization of an unusual deletion of the galactose-1-phosphate uridyl transferase (GALT) gene. Genet Med 8:635-40 [PubMed: 17079880]
- Coss KP, Byrne JC, Coman DJ, Adamczyk B, Abrahams JL, Saldova R, Brown AY, Walsh O, Hendroff U, Carolan C, Rudd PM, Treacy EP. IgG N-glycans as potential biomarkers for determining galactose tolerance in classical galactosaemia. Mol Genet Metab. 2012;105:212-20. [PubMed: 22133299]
- Coss KP, Doran PP, Owoeye C, Codd MB, Hamid N, Mayne PD, Crushell E, Knerr I, Monavari AA, Treacy EP. Classical galactosaemia in Ireland: incidence, complications and outcomes of treatment. J Inherit Metab Dis. 2013;36:21-7. [PubMed: 22870861]
- Crushell E, Chukwu J, Mayne P, Blatny J, Treacy EP. Negative screening tests in classical galactosaemia caused by S135L homozygosity. J Inherit Metab Dis. 2009;32:412-5. [PubMed: 19418241]
- Demirbas D, Huang X, Daesety V, Feenstra S, Haskovic M, Qi W, Gubbels CS, Hecht L, Levy HL, Waisbren SE, Berry GT. The ability of an LC-MS/MS-based erythrocyte GALT enzyme assay to predict the phenotype in subjects with GALT deficiency. Mol Genet Metab. 2019;126:368-76. [PubMed: 30718057]
- Doyle CM, Channon S, Orlowska D, Lee PJ. The neuropsychological profile of galactosaemia. J Inherit Metab Dis. 2010;33:603-9. [PubMed: 20607611]
- Elsas LJ 2nd, Lai K (1998) The molecular biology of galactosemia. Genet Med 1:40-8 [PubMed: 11261429]
- Elsas LJ, Webb AL, Langley SD (2002) Characterization of a carbohydrate response element regulating the gene for human galactose-1-phosphate uridyltransferase. Mol Genet Metab 76:287-96 [PubMed: 12208133]
- Frederick AB, Cutler DJ, Fridovich-Keil JL (2017) Rigor of non-dairy galactose restriction in early childhood, measured by retrospective survey, does not associate with severity of five long-term outcomes quantified in 231 children and adults with classic galactosemia. J Inherit Metab Dis. 40:813-21 [PMC free article: PMC5656392] [PubMed: 28695375]
- Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E. Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis. 2011;34:357-66. [PMC free article: PMC3063539] [PubMed: 20978943]
- Gubbels CS, Thomas CM, Wodzig WK, Olthaar AJ, Jaeken J, Sweep FC, Rubio-Gozalbo ME. FSH isoform pattern in classic galactosemia. J Inherit Metab Dis. 2011;34:387-90. [PMC free article: PMC3063565] [PubMed: 20814826]
- Gubbels CS, Welt CK, Dumoulin JC, Robben SG, Gordon CM, Dunselman GA, Rubio-Gozalbo ME, Berry GT. The male reproductive system in classic galactosemia: cryptorchidism and low semen volume. J Inherit Metab Dis. 2013;36:779-86. [PubMed: 23053469]
- Guerrero NV, Singh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ. Risk factors for premature ovarian failure in females with galactosemia. J Pediatr. 2000;137:833-41. [PubMed: 11113841]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549: 519-22. [PubMed: 28959963]
- Henderson H, Leisegang F, Brown R, Eley B. The clinical and molecular spectrum of galactosemia in patients from the Cape Town region of South Africa. BMC Pediatr. 2002;2:7. [PMC free article: PMC126267] [PubMed: 12350230]
- Hoffmann B, Dragano N, Schweitzer-Krantz S. Living situation, occupation and health-related quality of life in adult patients with classic galactosemia. J Inherit Metab Dis. 2012;35:1051-8. [PubMed: 22447152]
- Hoffmann B, Wendel U, Schweitzer-Krantz S. Cross-sectional analysis of speech and cognitive performance in 32 patients with classic galactosemia. J Inherit Metab Dis. 2011;34:421-7. [PubMed: 21347587]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Komrower GM. Galactosemia – thirty years on. The experience of a generation. J Inherit Metab Dis. 1982;5:96-104.
- Krabbi K, Uudelepp ML, Joost K, Zordania R, Õunap K. Long-term complications in Estonian galactosemia patients with a less strict lactose-free diet and metabolic control. Mol Genet Metab. 2011;103:249-53. [PubMed: 21501963]
- Lai K, Langley SD, Dembure PP, Hjelm LN, Elsas LJ 2nd (1998) Duarte allele impairs biostability of galactose-1-phosphate uridyltransferase in human lymphoblasts. Hum Mutat 11:28-38 [PubMed: 9450900]
- Lai K, Langley SD, Singh RH, Dembure PP, Hjelm LN, Elsas LJ 2nd (1996) A prevalent mutation for galactosemia among black Americans. J Pediatr 128:89-95 [PubMed: 8551426]
- Lai K, Tang M, Yin X, Klapper H, Wierenga K, Elsas L (2008) ARHI: A new target of galactose toxicity in classic galactosemia. Biosci Hypotheses. 1:263-71. [PMC free article: PMC2613282] [PubMed: 19122833]
- Langley SD, Lai K, Dembure PP, Hjelm LN, Elsas LJ (1997) Molecular basis for Duarte and Los Angeles variant galactosemia. Am J Hum Genet 60:366-72. [PMC free article: PMC1712399] [PubMed: 9012409]
- Lee PJ, Lilburn M, Wendel U, Schadewaldt P. A woman with untreated galactosaemia. Lancet. 2003;362:446. [PubMed: 12927432]
- Leslie ND, Immerman EB, Flach JE, Florez M, Fridovich-Keil JL, Elsas LJ (1992) The human galactose-1-phosphate uridyltransferase gene. Genomics 14:474-80 [PubMed: 1427861]
- Levy HL, Sepe SJ, Shih VE, Vawter GF, Klein JO. Sepsis due to Escherichia coli in neonates with galactosemia. N Engl J Med. 1977;297:823-5. [PubMed: 331112]
- Levy HL, Brown AE, Williams SE, de Juan E Jr. Vitreous hemorrhage as an ophthalmic complication of galactosemia. J Pediatr. 1996;129:922-5. [PubMed: 8969739]
- Malone JI, Diaz-Thomas A, Swan K. Problems with the newborn screen for galactosaemia. BMJ Case Rep. 2011;2011:bcr0120113769. [PMC free article: PMC3109760] [PubMed: 22693313]
- Mamsen LS, Kelsey TW, Ernst E, Macklon KT, Lund AM, Andersen CY. Cryopreservation of ovarian tissue may be considered in young girls with galactosemia. J Assist Reprod Genet. 2018;35:1209-17. [PMC free article: PMC6063818] [PubMed: 29804175]
- Menezo YJ, Lescaille M, Nicollet B, Servy EJ (2004) Pregnancy and delivery after stimulation with rFSH of a galatosemia patient suffering hypergonadotropic hypogonadism: case report. J Assist Reprod Genet 21:89-90. [PMC free article: PMC3455405] [PubMed: 15202737]
- National Newborn Screening and Genetics Resource Center. National newborn screening status report. 2014.
- Ng WG, Xu YK, Wong LJ, Kaufman FR, Buist NR, Donnell GN (2003) Two adult galactosaemia females with normal ovarian function and identical GALT mutations (Q188R/R333G). J Inherit Metab Dis 26:75-9 [PubMed: 12872845]
- Otaduy MC, Leite CC, Lacerda MT, Costa MO, Arita F, Prado E, Rosemberg S. Proton MR spectroscopy and imaging of a galactosemic patient before and after dietary treatment. Am J Neuroradiol. 2006;27:204-7. [PMC free article: PMC7976095] [PubMed: 16418384]
- Panis B, Bakker JA, Sels JP, Spaapen LJ, van Loon LJ, Rubio-Gozalbo ME (2006a) Untreated classical galactosemia patient with mild phenotype. Mol Genet Metab 89:277-9 [PubMed: 16621642]
- Panis B, Gerver WJ, Rubio-Gozalbo ME (2007) Growth in treated classical galactosemia patients. Eur J Pediatr 166:443-6 [PubMed: 17024348]
- Panis B, Vermeer C, van Kroonenburgh MJ, Nieman FH, Menheere PP, Spaapen LJ, Rubio-Gozalbo ME (2006b) Effect of calcium, vitamins K1 and D3 on bone in galactosemia. Bone 39:1123-9 [PubMed: 16782422]
- Potter NL, Nievergelt Y, Shriberg LD. Motor and speech disorders in classic galactosemia. JIMD Rep. 2013;11:31-41. [PMC free article: PMC3755563] [PubMed: 23546812]
- Robertson A, Singh RH (2000) Outcomes analysis of verbal dyspraxia in classic galactosemia. Genet Med 2:142-8 [PubMed: 11397328]
- Rubio-Agusti I, Carecchio M, Bhatia KP, Kojovic M, Parees I, Chandrashekar HS, Footitt EJ, Burke D, Edwards MJ, Lachmann RH, Murphy E. Movement disorders in adult patients with classical galactosemia. Mov Disord. 2013;28:804-10. [PubMed: 23400815]
- Rubio-Gozalbo ME, Gubbels C, Bakker J, Menherre P, Wodzig W, Land J. Gonadal function in’ male and female patients with classic galactosemia. Hum Reprod Update. 2010;16:177-88. [PubMed: 19793842]
- Rubio-Gozalbo ME, Haskovic M, Bosch AM, Burnyte B, Coelho AI, Cassiman D, Couce ML, Dawson C, Demirbas D, Derks T, Eyskens F, Forga MT, Grunewald S, Häberle J, Hochuli M, Hubert A, Huidekoper HH, Janeiro P, Kotzka J, Knerr I, Labrune P, Landau YE, Langendonk JG, Möslinger D, Müller-Wieland D, Murphy E, Õunap K, Ramadza D, Rivera IA, Scholl-Buergi S, Stepien KM, Thijs A, Tran C, Vara R, Visser G, Vos R, de Vries M, Waisbren SE, Welsink-Karssies MM, Wortmann SB, Gautschi M, Treacy EP, Berry GT. The natural history of classic galactosemia: lessons from the GalNet registry. Orphanet J Rare Dis. 2019;14:86. [PMC free article: PMC6486996] [PubMed: 31029175]
- Ryan EL, Lynch ME, Taddeo E, Gleason TJ, Epstein MP, Fridovich-Keil JL. Cryptic residual GALT activity is a potential modifier of scholastic outcome in school age children with classic galactosemia. J Inherit Metab Dis. 2013;36:1049-61. [PMC free article: PMC3657299] [PubMed: 23319291]
- Saudubray JM, van den Berghe G, Walter JH, eds. Inborn Metabolic Diseases: Diagnosis and Treatment. 5 ed. New York, NY: Springer. 2012.
- Schadewaldt P, Hoffmann B, Hammen HW, Kamp G, Schweitzer-Krantz S, Wendel U. Longitudinal assessment of intellectual achievement in patients with classical galactosemia. Pediatrics. 2010;125:e374-81. [PubMed: 20100763]
- Schadewaldt P, Kamalanathan L, Hammen HW, Wendel U (2004) Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol Genet Metab 81:31-44 [PubMed: 14728989]
- Shaw KA, Mulle JG, Epstein MP, Fridovich-Keil JL (2017) Gastrointestinal health in classic galactosemia. JIMD Rep. 33:27-32 [PMC free article: PMC5413454] [PubMed: 27363831]
- Shield JP, Wadsworth EJ, MacDonald A, Stephenson A, Tyfield L, Holton JB, Marlow N (2000) The relationship of genotype to cognitive outcome in galactosaemia. Arch Dis Child 83:248-50 [PMC free article: PMC1718484] [PubMed: 10952646]
- Takci S, Kadayifcilar S, Coskun T, Yigit S, Hismi B. A rare galactosemia complication: vitreous hemorrhage. JIMD Rep. 2012;5:89-93. [PMC free article: PMC3509908] [PubMed: 23430922]
- Tang M, Wierenga K, Elsas LJ, Lai K (2010) Molecular and biochemical characterization of human galactokinase and its small molecule inhibitors. Chem Biol Interact 188:376-85. [PMC free article: PMC2980576] [PubMed: 20696150]
- ten Hoedt AE, Maurice-Stam H, Boelen CC, Rubio-Gozalbo ME, van Spronsen FJ, Wijburg FA, Bosch AM, Grootenhuis MA. Parenting a child with phenylketonuria or galactosemia: implications for health-related quality of life. J Inherit Metab Dis. 2011;34:391-8. [PMC free article: PMC3063540] [PubMed: 21290186]
- Tyfield L, Reichardt J, Fridovich-Keil J, Croke DT, Elsas LJ 2nd, Strobl W, Kozak L, Coskun T, Novelli G, Okano Y, Zekanowski C, Shin Y, Boleda MD (1999) Classical galactosemia and mutations at the galactose-1-phosphate uridyl transferase (GALT) gene. Hum Mutat 13:417-30 [PubMed: 10408771]
- Tyfield LA (2000) Galactosemia and allelic variation at the galactose-1-phosphate uridyltransferase gene: a complex relationship between genotype and phenotype. Eur J Pediatr 159:S204-7 [PubMed: 11216901]
- van Erven B, Gubbels CS, van Golde RJ, Dunselman GA, Derhaag JG, de Wert G, Geraedts JP, Bosch AM, Treacy EP, Welt CK, Berry GT, Rubio-Gozalbo ME. Fertility preservation in female classic galactosemia patients. Orphanet J Rare Dis. 2013;8:107. [PMC free article: PMC3718676] [PubMed: 23866841]
- Wada Y, Kikuchi A, Arai-Ichinoi N, Sakamoto O, Takezawa Y, Iwasawa S, Niihori T, Nyuzuki H, Nakajima Y, Ogawa E, Ishige M, Hirai H, Sasai H, Fujiki R, Shirota M, Funayama R, Yamamoto M, Ito T, Ohara O, Nakayama K, Aoki Y, Koshiba S, Fukao T, Kure S. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet Med. 2019;21:1286-94. [PubMed: 30451973]
- Waggoner DD, Buist NR, Donnell GN (1990) Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis 13:802-18 [PubMed: 1706789]
- Waisbren SE, Potter NL, Gordon CM, Green RC, Greenstein P, Gubbels CS, Rubio-Gozalbo E, Schomer D, Welt C, Anastasoaie V, D'Anna K, Gentile J, Guo C-Y, Hecht L, Jackson R, Jansma BM, Li Y, Lip V, Miller DT, Murray M, Power L, Quinn N, Rohr F, Shen Y, Skinder-Meredith A, Timmers I, Tunick R, Wessel A, Wu B-L, Levy H, Elsas L, Berry GT. The adult galactosemic phenotype. J Inherit Metab Dis. 2012;35:279-86. [PMC free article: PMC3641771] [PubMed: 21779791]
- Walter JH, Fridovich-Keil JL. Galactosemia. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 72. New York, NY: McGraw-Hill. 2014.
- Webb AL, Singh RH, Kennedy MJ, Elsas LJ (2003) Verbal dyspraxia and galactosemia. Pediatr Res 53:396-402 [PubMed: 12595586]
- Welling L, Bernstein LE, Berry GT, Burlina AB, Eyskens F, Gautschi M, Grünewald S, Gubbels CS, Knerr I, Labrune P, van der Lee JH, MacDonald A, Murphy E, Portnoi PA, Õunap K, Potter NL, Rubio-Gozalbo ME, Spencer JB, Timmers I, Treacy EP, Van Calcar SC, Waisbren SE, Bosch AM, et al. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J Inherit Metab Dis. 2017;40:171-6. [PMC free article: PMC5306419] [PubMed: 27858262]
- Zlatunich CO, Packman S (2005) Galactosaemia: early treatment with an elemental formula. J Inherit Metab Dis 28:163-8 [PubMed: 15877205]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: February 4, 2000; Last Update: March 11, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Berry GT. Classic Galactosemia and Clinical Variant Galactosemia. 2000 Feb 4 [Updated 2021 Mar 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.