Summary
Clinical characteristics.
Rhabdoid tumor predisposition syndrome (RTPS) is characterized by a markedly increased risk for the development of rhabdoid tumors – rare and highly aggressive malignant tumors occurring predominantly in infants and children younger than age three years. Malignant rhabdoid tumors can occur in almost any anatomic location. They often occur in the central nervous system (i.e., atypical teratoid/rhabdoid tumor]); more than 50% occur in the cerebellum. Other common locations include extracranial malignant rhabdoid tumors (e.g., rhabdoid tumors of the head and neck, paravertebral muscles, liver, bladder, mediastinum, retroperitoneum, pelvis, and heart), rhabdoid tumor of the kidney, and possibly small-cell carcinoma of the ovary, hypercalcemic type (SCCOHT). More than 70% of individuals with RTPS present before age 12 months with synchronous tumors that exhibit aggressive clinical behavior.
Diagnosis/testing.
The diagnosis of RTPS is established in a proband with a rhabdoid tumor and/or a family history of rhabdoid tumor and/or multiple SMARCB1- or SMARCA4-deficient tumors (synchronous or metachronous), and a heterozygous disease-causing germline variant in SMARCB1 (RTPS1) or SMARCA4 (RTPS2) identified by molecular genetic testing.
Management.
Treatment of manifestations: Because of the rarity of RTPS, standards for management are evolving. Most individuals are treated using intensive multimodal therapeutic strategies – according to institutional preference – combining surgery, radiotherapy, and chemotherapy. The intensive multimodal treatment strategies required for clinically aggressive tumors in children with RTPS lead to a high rate of secondary complications. Consider risk-reducing treatment strategies (e.g., postpone or replace radiotherapy with high-dose chemotherapy or proton beam therapy; use targeted therapy concomitantly with, or before, standard chemotherapy).
Prevention of primary manifestations: Prophylactic risk-reducing bilateral salpingo-oophorectomy may be discussed following the end of family planning in women with SMARCA4-related RTPS because of the high risk of developing SCCOHT. The medical and ethical ramifications involved require an interdisciplinary approach including counseling and further research.
Surveillance: For all individuals with a disease-causing germline variant in SMARCB1 or SMARCA4 (regardless of age), whole-body MRI should be offered at diagnosis:
- Birth to age six months. Monthly (or at least every 2-3 months) thorough clinical examination including neurologic examination, ultrasound of the abdomen and neck, and head ultrasound or brain and spine MRI or whole-body MRI
- Age seven to 18 months. Every two to three months, thorough clinical examination including neurologic examination and ultrasound of the abdomen and neck. Consider brain and spine MRI as whole-body MRI resolution may not be sufficient for brain structures.
- Age 19 months to five years. Every three months, thorough clinical examination including neurologic examination, ultrasound of the abdomen and neck, and brain and spine MRI.
- After age five years. Every six months, thorough clinical examination including neurologic examination and annual whole-body MRI. Individuals with SMARCA4-related SCCOHT should have an abdominal and pelvic ultrasound every six months.
Evaluation of relatives at risk: It is appropriate to evaluate apparently asymptomatic older and younger at-risk relatives of an affected individual to identify as early as possible those who would benefit from prompt initiation of tumor surveillance.
Genetic counseling.
RTPS is inherited in an autosomal dominant fashion. The vast majority of individuals with SMARCB1-related RTPS have a de novo disease-causing SMARCB1 germline variant. Most reported individuals diagnosed with SMARCA4-related RTPS inherited a disease-causing variant from a parent without a history of a rhabdoid tumor or SCCOHT. Each child of an individual with a germline SMARCB1- or SMARCA4 disease-causing variant has a 50% chance of inheriting this variant. The penetrance of SMARCB1-related RTPS may be extremely high in individuals who inherit a SMARCB1 disease-causing variant. The penetrance of SMARCA4-related RTPS appears to be incomplete. The types of RTPS-related tumors vary among family members with the same disease-causing variant. Once the SMARCB1 or SMARCA4 disease-causing variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
Rhabdoid tumor predisposition syndrome (RTPS) should be suspected in an individual with any of the following clinical or laboratory features.
Clinical features. Any rhabdoid tumor with the following features is particularly suspicious:
- Congenital presentation (i.e., prenatal diagnosis or symptoms within the first 28 days of life)
- Early-onset rhabdoid tumor (age <12 months)
- Advanced stage of rhabdoid tumor at diagnosis (e.g., >M1 by Chang classification; Stage ≥II in extracranial malignant rhabdoid tumor [Harisiadis & Chang 1977])
- Synchronous rhabdoid tumors (>1 primary rhabdoid tumor)
- Family history of rhabdoid tumor, small-cell carcinoma of the ovary, hypercalcemic type, or other malignant entities such as cribriform neuroepithelial tumor, malignant peripheral nerve sheath tumor, myeloid sarcoma, epithelioid schwannoma, meningioma, benign myoepithelioma, chondrosarcoma, and/or ganglioglioma
- Family history of RTPS
Germline molecular genetic testing for RTPS is recommended in any individual with:
- A rhabdoid tumor (at any age), familial rhabdoid tumors, multifocal tumors, or congenital onset tumors;
- A SMARCB1-deficient tumor with a family history of rhabdoid tumor OR family history of nonspecified cancer in early childhood (age <5 years);
- A SMARCA4-deficient tumor with a family history of rhabdoid tumor OR family history of nonspecified cancer in early childhood (age <5 years).
Note: (1) It remains to be determined whether adult-onset rhabdoid tumors are caused by disease-causing germline variants in SMARCB1 or SMARCA4. (2) As morphologic rhabdoid features may not be present in all rhabdoid tumor biopsies because of inter- and intratumoral heterogeneity, any small blue round cell tumors in infants and young children should be evaluated for absence of nuclear SMARCB1 or SMARCA4 staining.
Laboratory features of tumor tissue
- Immunohistochemistry. Absence of SMARCB1 (INI-1) or SMARCA4 (BRG-1) staining indicating lack of functional protein in tumor tissue
- Molecular genetic testing. Somatic SMARCB1 or SMARCA4 disease-causing variants identified in a rhabdoid tumor. Note: Fresh-frozen tumor is preferable; formalin-fixed, paraffin-embedded samples may also be suitable.
Establishing the Diagnosis
There is currently no consensus regarding formal diagnostic criteria for RTPS.
The diagnosis of RTPS is established in any proband with both of the following:
- A rhabdoid tumor and/or a family history of rhabdoid tumor and/or multiple SMARCB1- or SMARCA4-deficient tumors (synchronous or metachronous) AND
- Identification of a disease-causing germline variant in SMARCB1 or SMARCA4 using molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include serial single-gene testing and use of a multigene panel.
- Serial single-gene testing may be considered in individuals with absence of SMARCB1 or SMARCA4 identified on tumor immunohistochemistry:
- Absence of SMARCB1. Sequence analysis and gene-targeted deletion/duplication analysis of SMARCB1 may be performed first.
- Absence of SMARCA4. Sequence analysis and gene-targeted deletion/duplication analysis of SMARCA4 may be performed first.
- A multigene panel that includes SMARCB1, SMARCA4, and other genes of interest (see Differential Diagnosis) may be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel provides the best opportunity to identify the genetic cause of the condition while limiting identification of disease-causing variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. (5) Multigene panel testing should include deletion/duplication analysis designed to detect smaller single-exon deletions and duplications and larger multiexon and whole gene deletions and duplications.
Table 1.
Molecular Genetic Testing Used in Rhabdoid Tumor Predisposition Syndrome
Clinical Characteristics
Clinical Description
Rhabdoid tumor predisposition syndrome (RTPS) is characterized by a markedly increased risk of developing rhabdoid tumors.
Rhabdoid tumors are rare and highly aggressive malignant tumors occurring predominantly in infants and children younger than age three years. The term rhabdoid is derived from the histologic resemblance of tumor cells to rhabdomyoblasts. Rhabdoid tumors are characterized by heaps of cells with an eccentric nucleus and prominent nucleoli, abundant cytoplasm with eosinophilic inclusion bodies, and distinct cellular membranes. Immunohistochemically, rhabdoid tumor cells are characterized by increased expression of vimentin (a nonspecific marker), epithelial membrane antigen, cytokeratins, and loss of SMARCB1 protein (a strong indicator for rhabdoid tumor) or, more rarely, of SMARCA4.
As morphologic rhabdoid features may not be present in all rhabdoid tumor biopsies because of inter- and intratumoral heterogeneity, any small blue round cell tumor in infants and young children should be evaluated for absence of nuclear SMARCB1 staining [Agaimy 2019].
Primary rhabdoid tumor locations include the following:
- Central nervous system: atypical teratoid/rhabdoid tumor (AT/RT); >50% are cerebellar.
- Head and neck, paravertebral muscles, liver, bladder, mediastinum, retroperitoneum, pelvis, and heart: extracranial malignant rhabdoid tumor (eMRT)
- Kidney: rhabdoid tumor of the kidney (RTK)
- Ovary: small-cell carcinoma of the ovary, hypercalcemic type (SCCOHT)
Rhabdoid tumors have been reported in nearly all anatomic locations [Brennan et al 2013, Frühwald et al 2020, Nemes et al 2021].
More than 70% of individuals with RTPS present before age 12 months with synchronous tumors that exhibit aggressive clinical behavior, often in one of the following clinical settings:
- Pre- or perinatally detected synchronous rhabdoid tumors [Negahban et al 2010, Fossey et al 2017, Nemes et al 2018, Schenone et al 2021]
- Infantile-onset or congenital rhabdoid tumor, presenting at a median age of five and a half months (range: prenatal to 60 months) compared to individuals with sporadic rhabdoid tumors (median age: 11.5 to 29.5 months; range: 1 day to 228 months) [Nemes et al 2018; Frühwald et al 2020; Nemes et al 2021; EU-RHAB ‒ Author, personal communication].Note: A bias toward increased molecular testing in younger individuals may confound the data.
- Synchronous (multiple primary) rhabdoid tumors. Ninety-one percent (10/11) of individuals with synchronous tumors demonstrated disease-causing germline variants [Nemes et al 2018]. Individuals with RTPS have a higher incidence of multiple rhabdoid tumors. Twenty-two of 90 individuals (24.5%) with RTPS in the EU-RHAB registry presented with synchronous tumors at diagnosis [EU-RHAB ‒ Author, personal communication].
- Family history of a rhabdoid tumor, epithelioid schwannoma, malignant peripheral nerve sheath tumor, myeloid sarcoma [Metts et al 2017], neuroblastoma [Coorens et al 2020], meningioma, benign myoepithelioma, chondrosarcoma, ganglioglioma, or cribriform neuroepithelial tumor [Bruggers et al 2011, Forest et al 2012, van den Munckhof et al 2012, Bosse et al 2014, Smith et al 2014, Evans et al 2018, Kehrer-Sawatzki et al 2018]. Rarely, AT/RTs occur in adult heterozygotes; for example, a sellar AT/RT-like tumor was described in a woman age 51 years whose daughter and brother had both died of malignant rhabdoid tumor [Voisin et al 2019].
- Family history of SCCOHT for SMARCA4-related RTPS (germline SMARCB1 disease-causing variants have not been reported in individuals with SCCOHT) [Schneppenheim et al 2010, Witkowski et al 2013, Witkowski et al 2016]Note: No individual with SCCOHT published to date developed a malignant rhabdoid tumor (MRT) during infancy. Apart from SCCOHT, truncating germline disease-causing variants of SMARCA4 have also been associated with undifferentiated uterine sarcomas and a single case of BRG1/SMARCA4-deficient lung carcinoma [Moes-Sosnowska et al 2015, Lavrut et al 2016, Witkowski et al 2017, Lin et al 2019, Connor et al 2020].
- Clinically aggressive rhabdoid tumors. Tumor progression at the time of follow up was identified in 84.5% (76/90) of individuals with RTPS [EU-RHAB ‒ Author, personal communication]. Progression occurred while on chemotherapy in 48% (35/73) of individuals with RTPS [Sredni & Tomita 2015; EU-RHAB ‒ Author, personal communication].
- Rhabdoid tumor and syndromic features suggestive of 22q11.2 distal deletion syndrome (OMIM 611867)
Prognosis. Individuals with RTPS potentially have a worse prognosis than those with a sporadic rhabdoid tumor, although long-term survival has been reported in some individuals [Kordes et al 2014, Seeringer et al 2014b, Nemes et al 2018, Frühwald et al 2020].
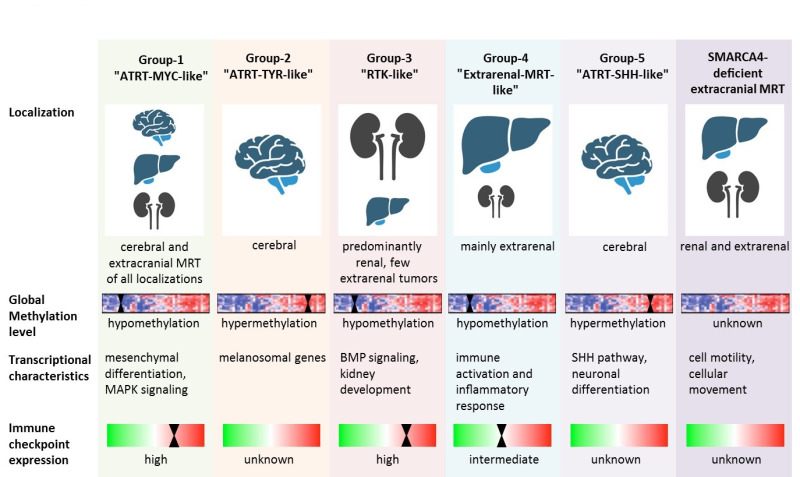
Pathogenesis. Various studies have revealed three molecular subgroups in AT/RT characterized by distinct transcriptional histopathologic and clinical characteristics: ATRT-TYR, ATRT-SHH, and ATRT-MYC, in accordance with overexpressed pathways or genes [Birks et al 2011, Birks et al 2013, Torchia et al 2015, Johann et al 2016, Torchia et al 2016, Ho et al 2020].
While these molecular groups are widely accepted in AT/RT, the molecular subgroups of eMRT are still evolving. Chun et al [2016] demonstrated two distinct molecular subgroups in eMRT (subgroup 1 and subgroup 2). Within subgroup 1, significantly overexpressed genes were linked to BMP signaling and differentiation. In subgroup 2, the most significantly overexpressed genes were linked to cell adhesion and migration, WNT signaling, and differentiation.
In a more recent integrative analysis of genomic, transcriptomic, and epigenomic profiles of 301 malignant rhabdoid tumors, five DNA methylation groups were identified based on anatomic site, SMARCB1 variants, gene expression pathways, DNA methylation-based pathway enrichment, and immune cell infiltration [Chun et al 2019]. Group 2 and group 5 of this integrative subgrouping correspond to the subgroups AT/RT-TYR and AT/RT-SHH as previously described in Johann et al [2016] and Ho et al [2020].
- Group 1 – AT/RT-MYC-like (ATRT-MYC and a subgroup of eMRT)
- Group 2 – AT/RT-TYR
- Group 3 – RTK-like
- Group 4 – Extrarenal MRT-like
- Group 5 – AT/RT-SHH
Notably, subgroup 2 (identified by differential gene expression analyses) largely corresponded to group 3 (RTK-like), while there was no clear equivalent for subgroup 1.
Groups 1, 3, and 4 (AT/RT-MYC-like, RTK-like, and extrarenal MRT-like) overexpressed HOX and other homeobox-containing genes involved in mesodermal development.
Groups 2 and 5 (AT/RT-TYR and AT/RT-SHH) largely corresponded to the respective AT/RT subgroups and were characterized by an expression of melanosomal features (AT/RT-TYR) and a proneural gene expression profile (AT/RT-SHH).
These results suggest that eMRTs share molecular features with AT/RT-MYC. Another feature distinguishing AT/RT-TYR and AT/RT-SHH from most other pediatric brain tumors is genome-wide hypermethylation – a characteristic that is not present in eMRT subgroups 1 and 2 and AT/RT-MYC.
Information on the specific molecular characteristics of SMARCA4-deficient eMRT has been sparse until very recently. A recent study shed light on the specific transcriptomic and DNA methylation characteristics of these entities [Andrianteranagna et al 2021]. While SMARCB1-deficient eMRT clustered together with AT/RT-MYC, the SMARCA4-deficient counterparts tended to form a separate cluster. Along a similar line, the transcriptomic characteristics of SMARCA4-deficient eMRT differed from SMARCB1-deficient eMRT. The molecular characteristics of the different eMRT subgroups are depicted in Figure 1.

Figure 1.
Overview of molecular features of the malignant rhabdoid tumor subgroups Reprinted from Nemes et al [2022]
Note: Current data suggest the value of subgroup determination for diagnostic and therapeutic decision making. A study by Brocks et al [2017] described an induction of cryptic transcription start sites (and thus putative neoantigens) following exposure to DNA demethylating agents. Whether this characteristic may also be found in a priori hypomethylated tumors remains to be studied.
Phenotype Correlations by Gene
SMARCA4. SCCOHT has been reported in individuals with SMARCA4-related RTPS and has not been reported in individuals with germline RTPS-associated SMARCB1 disease-causing variants.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Penetrance
SMARCB1. Penetrance of SMARCB1-related RTPS may be extremely high (>90% by age 5 years) [Holsten et al 2018, Nemes et al 2018]. However, these data may be based on selection bias, and larger series of systematically screened trios (parents and affected offspring) are needed to accurately define penetrance. Rarely a SMARCB1 disease-causing variant is inherited from an unaffected parent or a parent with late-onset or undiagnosed RTPS [Ammerlaan et al 2008]. Germline mosaicism may account for up to half of the families with sibs affected by RTPS.
SMARCA4. Even less is known about the penetrance of SMARCA4-related RTPS. Penetrance is incomplete, as most individuals with SMARCA4-related RTPS have inherited the disease-causing variant from an unaffected, healthy parent [Schneppenheim et al 2010; Hasselblatt et al 2014; Andrianteranagna et al 2021; Holdhof et al 2021; EU-RHAB ‒ Author, personal communication].
Nomenclature
RTPS may also be referred to as familial posterior fossa brain tumor syndrome.
RTPS1 refers to the predisposition associated with germline SMARCB1 disease-causing variants.
RTPS2, as initially described by Schneppenheim et al [2010], refers to the predisposition associated with disease-causing germline SMARCA4 variants, and could also be used to refer to SMARCA4-related cancers including SCCOHT.
Prevalence
Among newly diagnosed individuals with rhabdoid tumors, 25%-35% will have a disease-causing germline variant in SMARCB1 [Bourdeaut et al 2011, Eaton et al 2011, Hasselblatt et al 2014, Frühwald et al 2020, Nemes et al 2021].
The incidence of rhabdoid tumors may be estimated according to the following data:
- The age-standardized annual incidence rate is between 5 (eMRT) and 8.1 per million (AT/RT) in children younger than age one year and decreases to between 0.6 and 2.2 per million at ages one to four years [Brennan et al 2013, Dho et al 2015, German Childhood Cancer Registry 2019, Frühwald et al 2020, Nemes et al 2021].
- In the United States, annual incidence among children younger than age 15 years is 0.89 per million for AT/RT, 0.32 per million for eMRT, and 0.2 per million for RTK [Heck et al 2013, Nakata et al 2020].
Genetically Related (Allelic) Disorders
Other phenotypes caused by germline disease-causing variants in SMARCB1 and SMARCA4 are summarized in Table 2. Of note, rhabdoid tumors have not been observed in any of these phenotypes.
Table 2.
Allelic Disorders
Sporadic tumors occurring as single tumors in the absence of other findings of rhabdoid tumor predisposition syndrome (RTPS) may harbor a somatic disease-causing variant of SMARCB1 or SMARCA4 that is not present in the germline. In these circumstances predisposition to these tumors is not heritable. For more information, see Cancer and Benign Tumors.
Differential Diagnosis
Demonstration of loss of the SMARCB1 or SMARCA4 protein (in tumor tissue) as a result of inactivation or loss of one allele of SMARCB1 or SMARCA4 (tumor tissue and constitutional samples) may suggest the diagnosis of rhabdoid tumor predisposition syndrome (RTPS). For example, an individual with a constitutional deletion of SMARCB1 and an epithelioid sarcoma was reported by Le Loarer et al [2014]. In such cases the absence of a clinical and family history of rhabdoid tumor(s) distinguishes these individuals from those with RTPS.
Table 3.
Hereditary Disorders in the Differential Diagnosis of Rhabdoid Tumor Predisposition Syndrome
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with rhabdoid tumor predisposition syndrome (RTPS), the following are recommended:
- For all individuals (regardless of age), a whole-body MRI should be offered at diagnosis.
- Individuals who have not yet developed a rhabdoid tumor should be referred to a pediatric oncologist or tumor surveillance program.
- In those with a tumor, prior to planning therapy consider consulting a radiologist to assist in the selection and review of subsequent imaging, to evaluate the size and location of the primary tumor, and to evaluate for the presence of synchronous tumors and/or metastases (whole-body MRI).
- For individuals with atypical teratoid/rhabdoid tumor (AT/RT), examine cerebrospinal fluid and determine classification according to Chang staging [Harisiadis & Chang 1977].
- Refer to genetic counseling to inform affected individuals and their families about the nature, mode of inheritance, and implications of RTPS to facilitate medical and personal decision making.
Treatment of Manifestations
Because of the rarity of RTPS, standards for management are evolving. Most individuals are treated using intensive multimodal therapeutic strategies combining surgery, radiotherapy, and chemotherapy according to institutional preference:
- The Children's Oncology Group has employed a combination of surgery, two cycles of induction chemotherapy (cisplatin, cyclophosphamide, etoposide, vincristine, methotrexate), three cycles of high-dose chemotherapy (HDCT) with stem cell rescue (thiotepa, carboplatin) as consolidation therapy, and radiotherapy according to age and stage [Reddy et al 2020].
- The Dana-Farber Consortium has tested combination therapy with surgery, radiotherapy, and chemotherapy (vincristine, dactinomycin, cyclophosphamide, cisplatin, doxorubicin, temozolomide and intrathecal methotrexate, cytarabine, and hydrocortisone) [Chi et al 2009].
- The EU-RHAB registry recommends using combination therapy for rhabdoid tumors of any location (e.g., AT/RT, rhabdoid tumor of the kidney, extracranial malignant rhabdoid tumor), including gross total resection, conventional chemotherapy (vincristine, dactinomycin, cyclophosphamide, doxorubicin, ifosfamide, carboplatin, etoposide), intrathecal methotrexate, and permissive use of HDCT with stem cell rescue (carboplatin, thiotepa) and radiotherapy (in individuals age >18 months). The feasibility of intensive multimodal regimens even in the youngest individuals including those affected by RTPS has been demonstrated [Seeringer et al 2014a, Bartelheim et al 2016, Furtwängler et al 2018, Benesch et al 2020, Frühwald et al 2020, Nemes et al 2021].
- The Canadian Brain Tumour Consortium retrospectively evaluated children diagnosed with rhabdoid tumors between 1995 and 2007. Among 40 individuals, 22 received conventional chemotherapy and 18 received HDCT regimens; 15 received adjuvant radiation. Notably, six of 12 long-term survivors never received any radiotherapy [Lafay-Cousin et al 2012].
- Zaky et al [2014] evaluated the Head Start III experience for newly diagnosed individuals with AT/RT. Between 2003 and 2009, 19 individuals were treated with a combination of surgery and five courses of induction chemotherapy followed by consolidation with myeloablative chemotherapy and autologous hematopoietic progenitor cell rescue and radiotherapy according to age and stage. In five individuals, toxicity-related deaths occurred; ten individuals died as a result of disease progression. The three-year overall survival (OS) and event-free survival rates were 26±10% and 21±9%, respectively.
- Schrey et al [2016] summarized HDCT data by an individual pooled data analysis of 12 manuscripts and 389 publications including prospective and retrospective studies focused on the treatment of children diagnosed with AT/RT. Data of 332 individuals demonstrated an improved outcome in those treated with HDCT with stem cell rescue and radiotherapy.
- Fischer-Valuck et al [2017] evaluated data of 361 children diagnosed with AT/RT between 2004 and 2012. The five-year OS rate was 29.9%. For individuals with localized disease treated with multimodal therapy (surgery, chemotherapy, and radiotherapy), it was significantly higher, with a five-year OS rate of 46.8%. Individuals younger than age three years at diagnosis showed a significantly worse OS rate (5-year OS = 27.7%) compared to older individuals (5-year OS = 37.5%) and were also significantly less likely to receive multimodal therapy (specifically, the radiotherapy component). The authors suggest early radiotherapy as an important factor for long-term cure.
Note: RTPS most commonly affects infants; therapy presents a complex challenge because of the vulnerability of infants. The use of aggressive multimodal treatment on the developing nervous system and other organ systems in a young individual may profoundly affect neurodevelopmental outcome and lead to significant short- and long-term side effects. Intensive induction chemotherapy may often achieve a good response, and individuals may proceed with radiotherapy or (tandem) HDCT followed by autologous stem cell support.
The intensive multimodal treatment strategies required for clinically aggressive tumors in children with RTPS lead to a higher rate of secondary complications. Therapies and interventions that may prevent secondary complications include consideration of risk-reducing treatment strategies (e.g., postponing or replacing radiotherapy with HDCT or proton beam therapy; targeted therapy used concomitantly with – or before – standard chemotherapy). It remains to be determined whether a subgroup of children may be cured by surgery and chemotherapy alone, thus avoiding the potential severe side effects of radiotherapy to the developing brain.
Prevention of Primary Manifestations
Prophylactic risk-reducing bilateral salpingo-oophorectomy may be discussed following the end of family planning in women with SMARCA4-related RTPS because of the high risk of developing small-cell carcinoma of the ovary, hypercalcemic type (SCCOHT). The medical and ethical ramifications involved necessitate an interdisciplinary approach including counseling and further research [Berchuck et al 2015, Pejovic et al 2019].
Surveillance
Surveillance guidelines for individuals with RTPS have been provided by Teplick et al [2011], Foulkes et al [2017], and Frühwald et al [2021].
Birth to age six months. Monthly (or at least every 2-3 months) thorough clinical examination including neurologic examination, ultrasound of the abdomen and neck, and head ultrasound or brain and spine MRI or whole-body MRI (imaging modality is based on resources and need for anesthesia). Clinically suspicious regions should initially be evaluated by ultrasound. Note: This intensive surveillance may only be possible in a research setting; recommendations are based on the high risk of tumors within this age group.
Age seven months to 18 months. Every two to three months, thorough clinical examination including neurologic examination and ultrasound of the abdomen and neck; consider brain and spine MRI. Clinically suspicious regions should initially be evaluated by ultrasound. Note: Whole-body MRI resolution may not be sufficient for brain structures; MRI of the central nervous system will then need to be done separately.
Age 19 months to five years. Every three months, thorough clinical examination including neurologic examination, ultrasound of the abdomen and neck, and brain and spine MRI. Clinically suspicious regions should initially be evaluated by ultrasound.
After age five years the risk of developing a new rhabdoid tumor dramatically decreases [Eaton et al 2011]. It remains worthwhile, however, to screen individuals with RTPS for other manifestations (e.g., schwannomas, SCCOHT). A practical approach would include:
- Every six months, thorough clinical examination including neurologic examination;
- Annual whole-body MRI;
- Abdominal and pelvic ultrasound every six months in individuals with SMARCA4-related SCCOHT.
Note: (1) Current data do not allow for a determination of how long surveillance should continue. (2) Recommendations are subject to continuous updates; the authors recommend close monitoring of the evolving literature.
Agents/Circumstances to Avoid
Limit exposure to DNA-damaging agents including radiation (e.g., x-ray, CT, external beam radiotherapy), tobacco, UV light, and chemotherapy to minimize the lifetime risk of developing late-onset secondary cancers. Imaging tests utilizing radioactive compounds should be used only if absolutely necessary for essential health care. This recommendation is based on the increased risk of adverse effects in young developing children, not increased risk as a result of a SMARCA4 or SMARCB1 pathogenic variant.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of at-risk relatives of an affected individual by molecular genetic testing for the SMARCB1 or SMARCA4 disease-causing variant in the family.
- Early detection of individuals who are heterozygous for an SMARCB1 or SMARCA4 disease-causing variant allows prompt initiation of surveillance and treatment.
- Family members who have not inherited the disease-causing variant and their subsequent offspring have risks similar to the general population.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
The following clinical trials are currently recruiting unless otherwise indicated.
Table 4.
Overview of Clinical Trials in Pediatric Malignant Rhabdoid Tumors Including Rhabdoid Tumor Predisposition Syndrome
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Rhabdoid tumor predisposition syndrome (RTPS) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- The vast majority of individuals diagnosed with SMARCB1-related RTPS have the disorder as the result of a de novo germline disease-causing variant [Biegel et al 2014]. Rarely, a SMARCB1 disease-causing variant is inherited from an unaffected parent or a parent with late-onset or undiagnosed RTPS [Ammerlaan et al 2008].
- Most reported individuals diagnosed with SMARCA4-related RTPS inherited a disease-causing variant from a parent without a history of a rhabdoid tumor or small-cell carcinoma of the ovary, hypercalcemic type (SCCOHT) [Schneppenheim et al 2010; Hasselblatt et al 2014; Andrianteranagna et al 2021; Holdhof et al 2021; EU-RHAB ‒ Author, personal communication].
- If a proband appears to be the only affected family member (i.e., a simplex case), molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If a phenotypically healthy parent is found to have a SMARCB1 or SMARCA4 disease-causing germline variant, the parent should be offered surveillance.
- If the SMARCB1 or SMARCA4 disease-causing variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo disease-causing variant.
- The proband inherited a disease-causing variant from a parent with germline (or somatic and germline) mosaicism. Parental germline mosaicism in SMARCB1-related RTPS has been reported [Bruggers et al 2011, Eaton et al 2011, Biegel et al 2014, Gigante et al 2016]. Germline mosaicism may account for up to half of the families with sibs affected by RTPS.Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a disease-causing variant that is present in the germ cells only.
- The family history of most individuals with RTPS may appear to be negative because of failure to recognize the disorder in family members, reduced penetrance (highly likely in SMARCA4-related RTPS), or late onset in the affected parent. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the disease-causing variant identified in the proband.
Sibs of a proband. The risk of the sibs of a proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected and/or known to have the SMARCB1 or SMARCA4 disease-causing variant identified in the proband, the risk to the sibs of inheriting the variant is 50%.
- The penetrance of SMARCB1-related RTPS may be extremely high (e.g., >90% by age 5 years) in sibs who inherit a SMARCB1 disease-causing variant. Sibs may develop rhabdoid tumors and should undergo surveillance.
- Reduced penetrance is observed in SMARCA4-related RTPS. Sibs who inherit a SMARCA4 disease-causing variant may or may not develop rhabdoid tumors and should undergo surveillance.
- The types of RTPS-related tumors can vary among family members with the same disease-causing variant.
- If the SMARCB1 or SMARCA4 disease-causing variant identified in the proband cannot be detected in the constitutional (i.e., leukocyte) DNA of either parent, the recurrence risk to sibs is still greater than that of the general population because of the possibility of parental germline mosaicism [Bruggers et al 2011, Eaton et al 2011, Biegel et al 2014, Gigante et al 2016]. Germline mosaicism may account for up to half of the families with sibs affected by RTPS.
- If the parents have not been tested for the disease-causing variant identified in the proband but are clinically unaffected, sibs are still presumed to be at increased risk for RTPS because of the possibility of reduced penetrance in a heterozygous parent (highly likely in SMARCA4-related RTPS) or parental germline mosaicism.
Offspring of a proband. Each child of an individual with a germline SMARCB1 or SMARCA4 disease-causing variant has a 50% chance of inheriting the disease-causing variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has a germline SMARCB1 or SMARCA4 disease-causing variant, members of the parent's family may be at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Genetic cancer risk assessment and counseling. For a comprehensive description of the medical, psychosocial, and ethical ramifications of identifying at-risk individuals through cancer risk assessment with or without molecular genetic testing, see Cancer Genetics Risk Assessment and Counseling – Health Professional Version (part of PDQ®, National Cancer Institute).
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
High-risk pregnancies (i.e., those with a family history of RTPS). Once the SMARCB1 or SMARCA4 disease-causing variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Note: Rhabdoid tumors associated with RTPS may develop before birth; therefore, if the SMARCB1 or SMARCA4 disease-causing variant has been identified in the fetus, high-level ultrasound examination may be used to detect and identify prenatal manifestations of a primary tumor.
Low-risk pregnancies (i.e., those without a known family history of RTPS). If a primary tumor is detected on general prenatal ultrasound screening (and confirmed with high-level ultrasound), prenatal testing for a SMARCB1 or SMARCA4 disease-causing variant may be discussed. Note: Specific treatment for this group is currently not available, but interventions may be discussed; long-term survival has been reported in some affected individuals [Seeringer et al 2014b, Nemes et al 2018].
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- American Cancer SocietyPhone: 800-227-2345
- American Childhood Cancer OrganizationPhone: 855-858-2226
- CancerCarePhone: 800-813-4673Email: info@cancercare.org
- National Brain Tumor SocietyPhone: 617-924-9997Email: info@braintumor.org
- European Rhabdoid Registry (EU-RHAB)Stenglingstr. 2.Augsburg 86156GermanyPhone: 00498214004342Fax: 0049821400174243Email: eurhab@klinikum-augsburg.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Rhabdoid Tumor Predisposition Syndrome: Genes and Databases
Table B.
OMIM Entries for Rhabdoid Tumor Predisposition Syndrome (View All in OMIM)
Molecular Pathogenesis
Rhabdoid tumor predisposition syndrome (RTPS) is typically characterized by heterozygous germline disease-causing variants that predict inactivation of SMARCB1 (more commonly) or SMARCA4 (very rarely).
SMARCA4 with its ATPase activity is the catalytic subunit and SMARCB1 is a core protein of the SWI/SNF chromatin remodeling complex. SWI/SNF interacts with various pathways (p16-Rb pathway, Wnt/β-catenin pathway, sonic hedgehog signal pathway, polycomb pathway, MYCC, Aurora A) and affects many essential biological functions in developing organs, including cell cycle and cell differentiation, gene expression, and DNA repair [Biegel et al 2014, Kim & Roberts 2014, Kohashi & Oda 2017].
Mechanism of disease causation. Loss of function
Gene-specific laboratory technical considerations
- SMARCA4. By convention, disease-causing variants are numbered based on the sequence of the transcript encoding the longest isoform, which is transcript NM_001128849.3, comprising 36 exons. Reported disease-causing variants include nonsense and splice site variants and intragenic deletions that predict inactivation.
- SMARCB1. By convention, disease-causing variants are numbered based on the sequence of the transcript encoding the isoform a, which is transcript NM_003073.5, comprising nine exons. Reported disease-causing variants include nonsense and splice site variants, duplications, intragenic deletions, and exon and whole-gene deletions, which predict inactivation.
Cancer and Benign Tumors
Sporadic rhabdoid tumors may occur as single tumors in the absence of any other findings of RTPS and harbor somatic (acquired) SMARCB1 and/or SMARCA4 variants that are not present in the germline [Schneppenheim et al 2010, Biegel et al 2014]. In these circumstances predisposition to these tumors is not heritable. The routine application of immunohistochemistry to all neural tumors has identified other tumors with loss of SMARCA4 and/or SMARCB1 expression [Biegel et al 2014, Margol & Judkins 2014]. Whether SMARCB1 or SMARCA4 plays a role in development of these tumors is not known.
Chapter Notes
Author Notes
The European Rhabdoid Registry (EU-RHAB) was established in 2010 to define a standard of care for affected individuals with malignant rhabdoid tumors, to further the understanding of basic molecular mechanisms by coordinating the collection and analysis of biological materials in order to identify potential targets for pharmaceutical treatment, and eventually to shape the basis for Phase I/II trials.
Research projects
- SIOPE ATRT01 Study. An international prospective umbrella trial for children with atypical teratoid/rhabdoid tumors (AT/RT) including a randomized Phase III study evaluating the non-inferiority of three courses of high-dose chemotherapy compared to focal radiotherapy as consolidation therapy
- RTPS Project. "Families with rhabdoid tumor predisposition syndromes (RTPS1 and 2) – development of a clinical and human genetic concept"
- Relapse Project. "Mechanism of progression in malignant rhabdoid tumors"
Acknowledgments
MCF is supported by the "Deutsche Kinderkrebsstiftung" DKS 2020.10, "Deutsche Forschungsgemeinschaft" DFG FR 1516/4-1, and "Deutsche Krebshilfe" DKH 70113981. RS received grant support for infrastructure by the KinderKrebsInitiative Buchholz/Holm-Seppensen and "Deutsche Krebhilfe" DKH 70114040. PDJ is supported by the Else-Kroener-Fresenius Stiftung.
We thank P Neumayer and S Breitmoser for expert assistance in data acquisition, management, and analysis.
Author History
Karolina Nemes, MD, PhD (2017-present)
Susanne Bens, MD, PhD (2017-present)
Franck Bourdeaut, MD, PhD (2017-present)
Martin Hasselblatt, MD, Prof; University Hospital Münster (2017-2022)
Marcel Kool, PhD; German Cancer Consortium (2017-2022)
Pascal Johann, MD (2017-present)
Uwe Kordes, MD (2017-present)
Reinhard Schneppenheim, MD, PhD, Prof; University Medical Center Hamburg–Eppendorf (2017-2022)
Reiner Siebert, MD, Prof (2017-present)
Michael C Frühwald, MD, PhD, Prof (2017-present)
Revision History
- 12 May 2022 (sw) Comprehensive update posted live
- 7 December 2017 (sw) Review posted live
- 8 March 2017 (mcf) Original submission
References
Literature Cited
- Agaimy A. SWI/SNF complex-deficient soft tissue neoplasms: a pattern-based approach to diagnosis and differential diagnosis. Surg Pathol Clin. 2019;12:149–63. [PubMed: 30709441]
- Ammerlaan AC, Ararou A, Houben MP, Baas F, Tijssen CC, Teepen JL, Wesseling P, Hulsebos TJ. Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. Br J Cancer. 2008;98:474–9. [PMC free article: PMC2361463] [PubMed: 18087273]
- Andrianteranagna M, Cyrta J, Masliah-Planchon J, Nemes K, Corsia A, Leruste A, Holdhof D, Kordes U, Orbach D, Corradini N, Entz-Werle N, Pierron G, Castex MP, Brouchet A, Weingertner N, Ranchère D, Fréneaux P, Delattre O, Bush J, Leary A, Frühwald MC, Schüller U, Servant N, Bourdeaut F. SMARCA4-deficient rhabdoid tumours show intermediate molecular features between SMARCB1-deficient rhabdoid tumours and small cell carcinomas of the ovary, hypercalcaemic type. J Pathol. 2021;255:1–15. [PubMed: 33999421]
- Bartelheim K, Nemes K, Seeringer A, Kerl K, Buechner J, Boos J, Graf N, Dürken M, Gerss J, Hasselblatt M, Kortmann RD, Teichert von Luettichau I, Nagel I, Nygaard R, Oyen F, Quiroga E, Schlegel PG, Schmid I, Schneppenheim R, Siebert R, Solano-Paez P, Timmermann B, Warmuth-Metz M, Frühwald MC. Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med. 2016;5:1765–75. [PMC free article: PMC4884635] [PubMed: 27228363]
- Behnert A, Auber B, Steinemann D, Frühwald MC, Huisinga C, Hussein K, Kratz C, Ripperger T. KBG syndrome patient due to 16q24.3 microdeletion presenting with a paratesticular rhabdoid tumor: coincidence or cancer predisposition? Am J Med Genet A. 2018;176:1449–54. [PubMed: 29696793]
- Benesch M, Nemes K, Neumayer P, Hasselblatt M, Timmermann B, Bison B, Ebetsberger-Dachs G, Bourdeaut F, Dufour C, Biassoni V, Morales La Madrid A, Entz-Werle N, Laithier V, Quehenberger F, Weis S, Sumerauer D, Siebert R, Bens S, Schneppenheim R, Kool M, Modena P, Fouyssac F. C Frühwald M. Spinal cord atypical teratoid/rhabdoid tumors in children: clinical, genetic, and outcome characteristics in a representative European cohort. Pediatr Blood Cancer. 2020;67:e28022. [PubMed: 31571386]
- Berchuck A, Witkowski L, Hasselblatt M, Foulkes WD. Prophylactic oophorectomy for hereditary small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol Rep. 2015;12:20–2. [PMC free article: PMC4442656] [PubMed: 26076152]
- Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. 2014;166C:350–66. [PMC free article: PMC4516040] [PubMed: 25169151]
- Birks DK, Donson AM, Patel PR, Dunham C, Muscat A, Algar EM, Ashley DM, Kleinschmidt-Demasters BK, Vibhakar R, Handler MH, Foreman NK. High expression of BMP pathway genes distinguishes a subset of atypical teratoid/rhabdoid tumors associated with shorter survival. Neuro Oncol. 2011;13:1296–307. [PMC free article: PMC3223096] [PubMed: 21946044]
- Birks DK, Donson AM, Patel PR, Sufit A, Algar EM, Dunham C, Kleinschmidt-DeMasters BK, Handler MH, Vibhakar R, Foreman NK. Pediatric rhabdoid tumors of kidney and brain show many differences in gene expression but share dysregulation of cell cycle and epigenetic effector genes. Pediatr Blood Cancer. 2013;60:1095–102. [PMC free article: PMC4681512] [PubMed: 23382118]
- Bosse KR, Shukla AR, Pawel B, Chikwava KR, Santi M, Tooke L, Castagna K, Biegel JA, Bagatell R. Malignant rhabdoid tumor of the bladder and ganglioglioma in a 14 year-old male with a germline 22q11.2 deletion. Cancer Genet. 2014;207:415–9. [PMC free article: PMC7412592] [PubMed: 25018128]
- Bourdeaut F, Lequin D, Brugières L, Reynaud S, Dufour C, Doz F, André N, Stephan JL, Pérel Y, Oberlin O, Orbach D, Bergeron C, Rialland X, Fréneaux P, Ranchere D, Figarella-Branger D, Audry G, Puget S, Evans DG, Pinas JC, Capra V, Mosseri V, Coupier I, Gauthier-Villars M, Pierron G, Delattre O. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res. 2011;17:31–8. [PubMed: 21208904]
- Brennan B, Stiller C, Bourdeaut F. Extracranial rhabdoid tumours: what we have learned so far and future directions. Lancet Oncol. 2013;14:e329–36. [PubMed: 23816299]
- Brocks D, Schmidt CR, Daskalakis M, Jang HS, Shah NM, Li D, Li J, Zhang B, Hou Y, Laudato S, Lipka DB, Schott J, Bierhoff H, Assenov Y, Helf M, Ressnerova A, Islam MS, Lindroth AM, Haas S, Essers M, Imbusch CD, Brors B, Oehme I, Witt O, Lübbert M, Mallm JP, Rippe K, Will R, Weichenhan D, Stoecklin G, Gerhäuser C, Oakes CC, Wang T, Plass C. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet. 2017;49:1052–60. [PMC free article: PMC6005702] [PubMed: 28604729]
- Bruggers CS, Bleyl SB, Pysher T, Barnette P, Afify Z, Walker M, Biegel JA. Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer. 2011;56:1026–31. [PMC free article: PMC3210729] [PubMed: 20848638]
- Chi SN, Zimmerman MA, Yao X, Cohen KJ, Burger P, Biegel JA, Rorke-Adams LB, Fisher MJ, Janss A, Mazewski C, Goldman S, Manley PE, Bowers DC, Bendel A, Rubin J, Turner CD, Marcus KJ, Goumnerova L, Ullrich NJ, Kieran MW. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol. 2009;27:385–9. [PMC free article: PMC2645855] [PubMed: 19064966]
- Chun HE, Johann PD, Milne K, Zapatka M, Buellesbach A, Ishaque N, Iskar M, Erkek S, Wei L, Tessier-Cloutier B, Lever J, Titmuss E, Topham JT, Bowlby R, Chuah E, Mungall KL, Ma Y, Mungall AJ, Moore RA, Taylor MD, Gerhard DS, Jones SJM, Korshunov A, Gessler M, Kerl K, Hasselblatt M, Frühwald MC, Perlman EJ, Nelson BH, Pfister SM, Marra MA, Kool M. Identification and analyses of extra-cranial and cranial rhabdoid tumor molecular subgroups reveal tumors with cytotoxic T cell infiltration. Cell Rep. 2019;29:2338–54.e7. [PMC free article: PMC6905433] [PubMed: 31708418]
- Chun HE, Lim EL, Heravi-Moussavi A, Saberi S, Mungall KL, Bilenky M, Carles A, Tse K, Shlafman I, Zhu K, Qian JQ, Palmquist DL, He A, Long W, Goya R, Ng M, LeBlanc VG, Pleasance E, Thiessen N, Wong T, Chuah E, Zhao YJ, Schein JE, Gerhard DS, Taylor MD, Mungall AJ, Moore RA, Ma Y, Jones SJM, Perlman EJ, Hirst M, Marra MA. Genome-wide profiles of extra-cranial malignant rhabdoid tumors reveal heterogeneity and dysregulated developmental pathways. Cancer Cell. 2016;29:394–406. [PMC free article: PMC5094835] [PubMed: 26977886]
- Connor YD, Miao D, Lin DI, Hayne C, Howitt BE, Dalrymple JL, DeLeonardis KR, Hacker MR, Esselen KM, Shea M. Germline mutations of SMARCA4 in small cell carcinoma of the ovary, hypercalcemic type and in SMARCA4-deficient undifferentiated uterine sarcoma: clinical features of a single family and comparison of large cohorts. Gynecol Oncol. 2020;157:106–14. [PMC free article: PMC7887697] [PubMed: 31954538]
- Coorens THH, Farndon SJ, Mitchell TJ, Jain N, Lee S, Hubank M, Sebire N, Anderson J, Behjati S. Lineage-independent tumors in bilateral neuroblastoma. N Engl J Med. 2020;383:1860–5. [PMC free article: PMC7611571] [PubMed: 33211929]
- Dho YS, Kim SK, Cheon JE, Park SH, Wang KC, Lee JY, Phi JH. Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nerv Syst. 2015;31:1305–11. [PubMed: 25953096]
- Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011;56:7–15. [PMC free article: PMC3086793] [PubMed: 21108436]
- Errichiello E, Mustafa N, Vetro A, Notarangelo LD, de Jonge H, Rinaldi B, Vergani D, Giglio SR, Morbini P, Zuffardi O. SMARCA4 inactivating mutations cause concomitant Coffin-Siris syndrome, microphthalmia and small-cell carcinoma of the ovary hypercalcaemic type. J Pathol. 2017;243:9–15. [PMC free article: PMC5601212] [PubMed: 28608987]
- Evans DG, Bowers NL, Tobi S, Hartley C, Wallace AJ, King AT, Lloyd SKW, Rutherford SA, Hammerbeck-Ward C, Pathmanaban ON, Freeman SR, Ealing J, Kellett M, Laitt R, Thomas O, Halliday D, Ferner R, Taylor A, Duff C, Harkness EF, Smith MJ. Schwannomatosis: a genetic and epidemiological study. J Neurol Neurosurg Psychiatry. 2018;89:1215–19. [PubMed: 29909380]
- Fischer-Valuck BW, Chen I, Srivastava AJ, Floberg JM, Rao YJ, King AA, Shinohara ET, Perkins SM. Assessment of the treatment approach and survival outcomes in a modern cohort of patients with atypical teratoid rhabdoid tumors using the National Cancer Database. Cancer. 2017;123:682–7. [PubMed: 27861763]
- Forest F, David A, Arrufat S, Pierron G, Ranchere-Vince D, Stephan JL, Clemenson A, Delattre O, Bourdeaut F. Conventional chondrosarcoma in a survivor of rhabdoid tumor: enlarging the spectrum of tumors associated with SMARCB1 germline mutations. Am J Surg Pathol. 2012;36:1892–6. [PubMed: 23154773]
- Fossey M, Li H, Afzal S, Carret AS, Eisenstat DD, Fleming A, Hukin J, Hawkins C, Jabado N, Johnston D, Brown T, Larouche V, Scheinemann K, Strother D, Wilson B, Zelcer S, Huang A, Bouffet E, Lafay-Cousin L. Atypical teratoid rhabdoid tumor in the first year of life: the Canadian ATRT registry experience and review of the literature. J Neurooncol. 2017;132:155–62. [PubMed: 28102486]
- Foulkes WD, Kamihara J, Evans DGR, Brugières L, Bourdeaut F, Molenaar JJ, Walsh MF, Brodeur GM, Diller L. Cancer surveillance in Gorlin syndrome and rhabdoid tumor predisposition syndrome. Clin Cancer Res. 2017;23:e62–e67. [PMC free article: PMC7309678] [PubMed: 28620006]
- Frühwald MC, Hasselblatt M, Nemes K, Bens S, Steinbügl M, Johann PD, Kerl K, Hauser P, Quiroga E, Solano-Paez P, Biassoni V, Gil-da-Costa MJ, Perek-Polnik M, van de Wetering M, Sumerauer D, Pears J, Stabell N, Holm S, Hengartner H, Gerber NU, Grotzer M, Boos J, Ebinger M, Tippelt S, Paulus W, Furtwängler R. Hernáiz- Driever P, Reinhard H, Rutkowski S, Schlegel PG, Schmid I, Kortmann RD,Timmermann B, Warmuth-Metz M, Kordes U, Gerss J, Nysom K, Schneppenheim R, Siebert R, Kool M, Graf N. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol. 2020;22:1006–17. [PMC free article: PMC7339901] [PubMed: 31883020]
- Frühwald MC, Nemes K, Boztug H, Cornips MCA, Evans DG, Farah R, Glentis S, Jorgensen M, Katsibardi K, Hirsch S, Jahnukainen K, Kventsel I, Kerl K, Kratz CP, Pajtler KW, Kordes U, Ridola V, Stutz E, Bourdeaut F. Current recommendations for clinical surveillance and genetic testing in rhabdoid tumor predisposition: a report from the SIOPE Host Genome Working Group. Fam Cancer. 2021;20:305–16. [PMC free article: PMC8484234] [PubMed: 33532948]
- Furtwängler R, Kager L, Melchior P, Rübe C, Ebinger M, Nourkami-Tutdibi N, Niggli F, Warmann S, Hubertus J, Amman G, Leuschner I, Vokuhl C, Graf N, Frühwald MC. High-dose treatment for malignant rhabdoid tumor of the kidney: no evidence for improved survival-The Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) experience. Pediatr Blood Cancer. 2018:65. [PubMed: 28843054]
- German Childhood Cancer Registry. Annual report. 2019. Available online. Accessed 5-25-22.
- Gigante L, Paganini I, Frontali M, Ciabattoni S, Sangiuolo FC, Papi L. Rhabdoid tumor predisposition syndrome caused by SMARCB1 constitutional deletion: prenatal detection of new case of recurrence in siblings due to gonadal mosaicism. Fam Cancer. 2016;15:123–6. [PubMed: 26342593]
- Gossai N, Biegel JA, Messiaen L, Berry SA, Moertel CL. Report of a patient with a constitutional missense mutation in SMARCB1, Coffin-Siris phenotype, and schwannomatosis. Am J Med Genet A. 2015;167A:3186–91. [PubMed: 26364901]
- Harisiadis L, Chang CH. Medulloblastoma in children: a correlation between staging and results of treatment. Int J Radiat Oncol Biol Phys. 1977;2:833–41. [PubMed: 597384]
- Hasselblatt M, Nagel I, Oyen F, Bartelheim K, Russell RB, Schüller U, Junckerstorff R, Rosenblum M, Alassiri AH, Rossi S, Schmid I, Gottardo NG, Toledano H, Viscardi E, Balbin M, Witkowski L, Lu Q, Betts MJ, Foulkes WD, Siebert R, Frühwald MC, Schneppenheim R. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014;128:453–6. [PubMed: 25060813]
- Hasselblatt M, Thomas C, Federico A, Nemes K, Johann PD, Bison B, Bens S, Dahlum S, Kordes U, Redlich A, Lessel L, Pajtler KW, Mawrin C, Schüller U, Nolte K, Kramm CM, Hinz F, Sahm F, Giannini C, Penkert J, Kratz CP, Pfister SM, Siebert R, Paulus W, Kool M, Frühwald MC. SMARCB1-deficient and SMARCA4-deficient malignant brain tumors with complex copy number alterations and TP53 mutations may represent the first clinical manifestation of Li-Fraumeni syndrome. Am J Surg Pathol. 2022. Epub ahead of print. [PubMed: 35446794]
- Heck JE, Lombardi CA, Cockburn M, Meyers TJ, Wilhelm M, Ritz B. Epidemiology of rhabdoid tumors of early childhood. Pediatr Blood Cancer. 2013;60:77–81. [PMC free article: PMC3399923] [PubMed: 22434719]
- Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Frühwald M, Hasselblatt M, Bourdeaut F, Williamson D, Huang A, Kool M. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol. 2020;22:613–24. [PMC free article: PMC7229260] [PubMed: 31889194]
- Holdhof D, Johann PD, Spohn M, Bockmayr M, Safaei S, Joshi P, Masliah-Planchon J, Ho B, Andrianteranagna M, Bourdeaut F, Huang A, Kool M, Upadhyaya SA, Bendel AE, Indenbirken D, Foulkes WD, Bush JW, Creytens D, Kordes U, Frühwald MC, Hasselblatt M, Schüller U. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathol. 2021;141:291–301. [PMC free article: PMC7847432] [PubMed: 33331994]
- Holsten T, Bens S, Oyen F, Nemes K, Hasselblatt M, Kordes U, Siebert R, Frühwald MC, Schneppenheim R, Schüller U. Germline variants in SMARCB1 and other members of the BAF chromatin-remodeling complex across human disease entities: a meta-analysis. Eur J Hum Genet. 2018;26:1083–93. [PMC free article: PMC6057970] [PubMed: 29706634]
- Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, Jones DT, Sturm D, Hermann C, Segura Wang M, Korshunov A, Rhyzova M, Gröbner S, Brabetz S, Chavez L, Bens S, Gröschel S, Kratochwil F, Wittmann A, Sieber L, Geörg C, Wolf S, Beck K, Oyen F, Capper D, van Sluis P, Volckmann R, Koster J, Versteeg R, von Deimling A, Milde T, Witt O, Kulozik AE, Ebinger M, Shalaby T, Grotzer M, Sumerauer D, Zamecnik J, Mora J, Jabado N, Taylor MD, Huang A, Aronica E, Bertoni A, Radlwimmer B, Pietsch T, Schüller U, Schneppenheim R, Northcott PA, Korbel JO, Siebert R, Frühwald MC, Lichter P, Eils R, Gajjar A, Hasselblatt M, Pfister SM, Kool M. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016;29:379–93. [PubMed: 26923874]
- Kehrer-Sawatzki H, Kordes U, Seiffert S, Summerer A, Hagel C, Schüller U, Farschtschi S, Schneppenheim R, Bendszus M, Godel T, Mautner VF. Co-occurrence of schwannomatosis and rhabdoid tumor predisposition syndrome 1. Mol Genet Genomic Med. 2018;6:627–37. [PMC free article: PMC6081224] [PubMed: 29779243]
- Kim KH, Roberts CW. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 2014;207:365–72. [PMC free article: PMC4195815] [PubMed: 24853101]
- Kohashi K, Oda Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 2017;108:547–52. [PMC free article: PMC5406539] [PubMed: 28109176]
- Kordes U, Bartelheim K, Modena P, Massimino M, Biassoni V, Reinhard H, Hasselblatt M, Schneppenheim R, Frühwald MC. Favorable outcome of patients affected by rhabdoid tumors due to rhabdoid tumor predisposition syndrome (RTPS). Pediatr Blood Cancer. 2014;61:919–21. [PubMed: 24123847]
- Lafay-Cousin L, Hawkins C, Carret AS, Johnston D, Zelcer S, Wilson B, Jabado N, Scheinemann K, Eisenstat D, Fryer C, Fleming A, Mpofu C, Larouche V, Strother D, Bouffet E, Huang A. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer. 2012;48:353–9. [PubMed: 22023887]
- Lavrut PM, Le Loarer F, Normand C, Grosos C, Dubois R, Buenerd A, Conter C, Dijoud F, Blay JY, Collardeau-Frachon S. Small cell carcinoma of the ovary, hypercalcemic type: report of a bilateral case in a teenager associated with SMARCA4 germline mutation. Pediatr Dev Pathol. 2016;19:56–60. [PubMed: 26230154]
- Le Loarer F, Zhang L, Fletcher CD, Ribeiro A, Singer S, Italiano A, Neuville A, Houlier A, Chibon F, Coindre JM, Antonescu CR. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosomes Cancer. 2014;53:475–86. [PMC free article: PMC4226650] [PubMed: 24585572]
- Li D, Ahrens-Nicklas RC, Baker J, Bhambhani V, Calhoun A, Cohen JS, Deardorff MA, Fernández-Jaén A, Kamien B, Jain M, Mckenzie F, Mintz M, Motter C, Niles K, Ritter A, Rogers C, Roifman M, Townshend S, Ward-Melver C, Schrier Vergano SA. The variability of SMARCA4-related Coffin-Siris syndrome: Do nonsense candidate variants add to milder phenotypes? Am J Med Genet A. 2020;182:2058–67. [PubMed: 32686290]
- Lin DI, Allen JM, Hecht JL, Killian JK, Ngo NT, Edgerly C, Severson EA, Ali SM, Erlich RL, Ramkissoon SH, Vergilio JA, Ross JS, Elvin JA. SMARCA4 inactivation defines a subset of undifferentiated uterine sarcomas with rhabdoid and small cell features and germline mutation association. Mod Pathol. 2019;32:1675–87. [PubMed: 31190001]
- Margol AS, Judkins AR. Pathology and diagnosis of SMARCB1-deficient tumors. Cancer Genet. 2014;207:358–64. [PubMed: 25246033]
- Metts JL, Park SI, Soares BP, Fong C, Biegel JA, Goldsmith KC. Concurrent myeloid sarcoma, atypical teratoid/rhabdoid tumor, and hypereosinophilia in an infant with a germline SMARCB1 mutation. Pediatr Blood Cancer. 2017:64. [PubMed: 28111898]
- Moes-Sosnowska J, Szafron L, Nowakowska D, Dansonka-Mieszkowska A, Budzilowska A, Konopka B, Plisiecka-Halasa J, Podgorska A, Rzepecka IK, Kupryjanczyk J. Germline SMARCA4 mutations in patients with ovarian small cell carcinoma of hypercalcemic type. Orphanet J Rare Dis. 2015;10:32. [PMC free article: PMC4365965] [PubMed: 25886974]
- Nakata K, Colombet M, Stiller CA, Pritchard-Jones K, Steliarova-Foucher E. Incidence of childhood renal tumours: an international population-based study. Int J Cancer. 2020;147:3313–27. [PMC free article: PMC7689773] [PubMed: 32902866]
- Negahban S, Ahmadi N, Oryan A, Khojasteh HN, Aledavood A, Soleimanpour H, Mohammadianpanah M, Oschlies I, Gesk S, Siebert R, Daneshbod K, Daneshbod Y. Primary bilateral Burkitt lymphoma of the lactating breast: a case report and review of the literature. Mol Diagn Ther. 2010;14:243–50. [PubMed: 20799767]
- Nemes K, Bens S, Kachanov D, Teleshova M, Hauser P, Simon T, Tippelt S, Woessmann W, Beck O, Flotho C, Grigull L, Driever PH, Schlegel PG, Khurana C, Hering K, Kolb R, Leipold A, Abbink F, Gil-Da-Costa MJ, Benesch M, Kerl K, Lowis S, Marques CH, Graf N, Nysom K, Vokuhl C, Melchior P, Kröncke T, Schneppenheim R, Kordes U, Gerss J, Siebert R, Furtwängler R, Frühwald MC. Clinical and genetic risk factors define two risk groups of extracranial malignant rhabdoid tumours (eMRT/RTK). Eur J Cancer. 2021;142:112–22. [PubMed: 33249395]
- Nemes K, Clément N, Kachanov D, Bens S, Hasselblatt M, Timmermann B, Schneppenheim R, Gerss J, Siebert R, Furtwängler R, Bourdeaut F, Frühwald MC, et al. The extraordinary challenge of treating patients with congenital rhabdoid tumors--a collaborative European effort. Pediatr Blood Cancer. 2018;65:e26999. [PubMed: 29418059]
- Nemes K, Frühwald MC. Emerging therapeutic targets for the treatment of malignant rhabdoid tumors. Expert Opin Ther Targets. 2018;22:365–79. [PubMed: 29528755]
- Nemes K, Johann PD, Tüchert S, Melchior P, Vokuhl C, Siebert R, Furtwängler R, Frühwald MC. Current and emerging therapeutic approaches for extracranial malignant rhabdoid tumors. Cancer Manag Res. 2022;14:479–98. [PMC free article: PMC8841298] [PubMed: 35173482]
- Pejovic T, McCluggage WG, Krieg AJ, Xu F, Lee DM, Witkowski L, Foulkes WD. The dilemma of early preventive oophorectomy in familial small cell carcinoma of the ovary of hypercalcemic type. Gynecol Oncol Rep. 2019;28:47–49. [PMC free article: PMC6402228] [PubMed: 30886884]
- Prasad RN, Gardner UG, Yaney A, Prevedello DM, Koboldt DC, Thomas DL, Mardis ER, Palmer JD. Germline BAP1 mutation in a family with multi-generational meningioma with rhabdoid features: a case series and literature review. Front Oncol. 2021;11:721712. [PMC free article: PMC8421801] [PubMed: 34504799]
- Reddy AT, Strother DR, Judkins AR, Burger PC, Pollack IF, Krailo MD, Buxton AB, Williams-Hughes C, Fouladi M, Mahajan A, Merchant TE, Ho B, Mazewski CM, Lewis VA, Gajjar A, Vezina LG, Booth TN, Parsons KW, Poss VL, Zhou T, Biegel JA, Huang A. Efficacy of high-dose chemotherapy and three-dimensional conformal radiation for atypical teratoid/rhabdoid tumor: a report from the Children's Oncology Group Trial ACNS0333. J Clin Oncol. 2020;38:1175–85. [PMC free article: PMC7145589] [PubMed: 32105509]
- Schenone CV, King A, Castro E, Ketwaroo P, Donepudi R, Sanz-Cortes M. Prenatal detection of disseminated extrarenal malignant rhabdoid tumor with placental metastases. Ultrasound Obstet Gynecol. 2021;57:1008–10. [PubMed: 32621313]
- Schneppenheim R, Frühwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, Kreuz M, Leuschner I, Martin Subero JI, Obser T, Oyen F, Vater I, Siebert R. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86:279–84. [PMC free article: PMC2820190] [PubMed: 20137775]
- Schrey D, Carceller Lechón F, Malietzis G, Moreno L, Dufour C, Chi S, Lafay-Cousin L, von Hoff K, Athanasiou T, Marshall LV, Zacharoulis S. Multimodal therapy in children and adolescents with newly diagnosed atypical teratoid rhabdoid tumor: individual pooled data analysis and review of the literature. J Neurooncol. 2016;126:81–90. [PubMed: 26608522]
- Seeringer A, Bartelheim K, Kerl K, Hasselblatt M, Leuschner I, Rutkowski S, Timmermann B, Kortmann RD, Koscielniak E, Schneppenheim R, Warmuth-Metz M, Gerss J, Siebert R, Graf N, Boos J, Frühwald MC. Feasibility of intensive multimodal therapy in infants affected by rhabdoid tumors - experience of the EU-RHAB registry. Klin Padiatr. 2014a;226:143–8. [PubMed: 24633978]
- Seeringer A, Reinhard H, Hasselblatt M, Schneppenheim R, Siebert R, Bartelheim K, Leuschner I, Frühwald MC. Synchronous congenital malignant rhabdoid tumor of the orbit and atypical teratoid/rhabdoid tumor--feasibility and efficacy of multimodal therapy in a long-term survivor. Cancer Genet. 2014b;207:429–33. [PubMed: 25262118]
- Smith MJ, Wallace AJ, Bowers NL, Eaton H, Evans DG. SMARCB1 mutations in schwannomatosis and genotype correlations with rhabdoid tumors. Cancer Genet. 2014;207:373–8. [PubMed: 24933152]
- Sredni ST, Tomita T. Rhabdoid tumor predisposition syndrome. Pediatr Dev Pathol. 2015;18:49–58. [PubMed: 25494491]
- Teplick A, Kowalski M, Biegel JA, Nichols KE. Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr. 2011;170:285–94. [PMC free article: PMC3086787] [PubMed: 21210147]
- Torchia J, Golbourn B, Feng S, Ho KC, Sin-Chan P, Vasiljevic A, Norman JD, Guilhamon P, Garzia L, Agamez NR, Lu M, Chan TS, Picard D, de Antonellis P, Khuong-Quang DA, Planello AC, Zeller C, Barsyte-Lovejoy D, Lafay-Cousin L, Letourneau L, Bourgey M, Yu M, Gendoo DM, Dzamba M, Barszczyk M, Medina T, Riemenschneider AN, Morrissy AS, Ra YS, Ramaswamy V, Remke M, Dunham CP, Yip S, Ng HK, Lu JQ, Mehta V, Albrecht S, Pimentel J, Chan JA, Somers GR, Faria CC, Roque L, Fouladi M, Hoffman LM, Moore AS, Wang Y, Choi SA, Hansford JR, Catchpoole D, Birks DK, Foreman NK, Strother D, Klekner A, Bognár L, Garami M, Hauser P, Hortobágyi T, Wilson B, Hukin J, Carret AS, Van Meter TE, Hwang EI, Gajjar A, Chiou SH, Nakamura H, Toledano H, Fried I, Fults D, Wataya T, Fryer C, Eisenstat DD, Scheinemann K, Fleming AJ, Johnston DL, Michaud J, Zelcer S, Hammond R, Afzal S, Ramsay DA, Sirachainan N, Hongeng S, Larbcharoensub N, Grundy RG, Lulla RR, Fangusaro JR, Druker H, Bartels U, Grant R, Malkin D, McGlade CJ, Nicolaides T, Tihan T, Phillips J, Majewski J, Montpetit A, Bourque G, Bader GD, Reddy AT, Gillespie GY, Warmuth-Metz M, Rutkowski S, Tabori U, Lupien M, Brudno M, Schüller U, Pietsch T, Judkins AR, Hawkins CE, Bouffet E, Kim SK, Dirks PB, Taylor MD, Erdreich-Epstein A, Arrowsmith CH, De Carvalho DD, Rutka JT, Jabado N, Huang A. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell. 2016;30:891–908. [PMC free article: PMC5500911] [PubMed: 27960086]
- Torchia J, Picard D, Lafay-Cousin L, Hawkins CE, Kim SK, Letourneau L, Ra YS, Ho KC, Chan TS, Sin-Chan P, Dunham CP, Yip S, Ng HK, Lu JQ, Albrecht S, Pimentel J, Chan JA, Somers GR, Zielenska M, Faria CC, Roque L, Baskin B, Birks D, Foreman N, Strother D, Klekner A, Garami M, Hauser P, Hortobágyi T, Bognár L, Wilson B, Hukin J, Carret AS, Van Meter TE, Nakamura H, Toledano H, Fried I, Fults D, Wataya T, Fryer C, Eisenstat DD, Scheineman K, Johnston D, Michaud J, Zelcer S, Hammond R, Ramsay DA, Fleming AJ, Lulla RR, Fangusaro JR, Sirachainan N, Larbcharoensub N, Hongeng S, Barakzai MA, Montpetit A, Stephens D, Grundy RG, Schüller U, Nicolaides T, Tihan T, Phillips J, Taylor MD, Rutka JT, Dirks P, Bader GD, Warmuth-Metz M, Rutkowski S, Pietsch T, Judkins AR, Jabado N, Bouffet E, Huang A. Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol. 2015;16:569–82. [PubMed: 25882982]
- van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJ. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics. 2012;13:1–7. [PubMed: 22038540]
- Voisin MR, Ovenden C, Tsang DS, Gupta AA, Huang A, Gao AF, Diamandis P, Almeida JP, Gentili F. Atypical teratoid/rhabdoid sellar tumor in an adult with a familial history of a germline SMARCB1 mutation: case report and review of the literature. World Neurosurg. 2019;127:336–45. [PubMed: 31004861]
- Witkowski L, Donini N, Byler-Dann R, Knost JA, Albrecht S, Berchuck A, McCluggage WG, Hasselblatt M, Foulkes WD. The hereditary nature of small cell carcinoma of the ovary, hypercalcemic type: two new familial cases. Fam Cancer. 2017;16:395–9. [PMC free article: PMC5487815] [PubMed: 27866340]
- Witkowski L, Goudie C, Ramos P, Boshari T, Brunet JS, Karnezis AN, Longy M, Knost JA, Saloustros E, McCluggage WG, Stewart CJR, Hendricks WPD, Cunliffe H, Huntsman DG, Pautier P, Levine DA, Trent JM, Berchuck A, Hasselblatt M, Foulkes WD. The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol. 2016;141:454–60. [PMC free article: PMC6876126] [PubMed: 26975901]
- Witkowski L, Lalonde E, Zhang J, Albrecht S, Hamel N, Cavallone L, May ST, Nicholson JC, Coleman N, Murray MJ, Tauber PF, Huntsman DG, Schönberger S, Yandell D, Hasselblatt M, Tischkowitz MD, Majewski J, Foulkes WD. Familial rhabdoid tumour "avant la letter"--from pathology review to exome sequencing and back again. J Pathol. 2013;231:35–43. [PubMed: 23775540]
- Zaky W, Dhall G, Ji L, Haley K, Allen J, Atlas M, Bertolone S, Cornelius A, Gardner S, Patel R, Pradhan K, Shen V, Thompson S, Torkildson J, Sposto R, Finlay JL. Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: the Head Start III experience. Pediatr Blood Cancer. 2014;61:95–101. [PubMed: 23934933]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: December 7, 2017; Last Update: May 12, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Nemes K, Bens S, Bourdeaut F, et al. Rhabdoid Tumor Predisposition Syndrome. 2017 Dec 7 [Updated 2022 May 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.