3.1. CANDIDATE SUBSTANCE INITIAL SCREENING
Candidate substances for ETAP are initially screened for any mammalian in vivo repeated dose toxicity studies using a search of the US EPA ToxVal database (ToxValDB)3. If no suitable studies are identified from the ToxValDB, then a SEM is initiated using the methods published by Thayer and colleagues and described below to confirm or refute the absence of studies (Thayer et al. 2022a; Thayer et al. 2022b). Only substances that have no apparent publicly available mammalian in vivo repeated dose toxicity studies or suitable human evidence are further considered for an ETAP.
3.2. SYSTEMATIC EVIDENCE MAP DEVELOPMENT
3.2.1. POPULATIONS, EXPOSURES, COMPARATORS, AND OUTCOME (PECO) CRITERIA
PECO criteria (Morgan et al. 2018; Thayer et al. 2022a; Thayer et al. 2022b) are used to focus the research questions, search terms, and inclusion/exclusion parameters in a systematic review (Table 3-1). Studies that did not meet the PECO criteria but contain relevant supporting information are categorized (or “tagged”) as potentially relevant supplemental material during the literature screening process (Table 3-2).
Table 3-1
Summary of PECO elements and associated evidence is as described in Thayer et al. 2022.
Table 3-2
Major categories of potentially relevant supplemental material.
3.2.2. LITERATURE SEARCH STRATEGIES
3.2.2.1. Database Search Term Development
The literature search focuses on the substance identifiers (name, synonyms, or trade names) with no date or language limits. Substance synonyms are identified by using synonyms in the EPA’s CompTox Chemicals Dashboard4 indicated as “valid” or “good”. The preferred chemical name, Chemical Abstracts Service Registry Number (CASRN), DSSTox substance identifier (DTXSID), and synonyms are used by EPA information specialists to develop search strategies tailored to each of the databases listed below.
3.2.2.2. Database Searches
The three databases listed below are queried for literature containing the chemical search terms, and all retrieved records are stored in the Health and Environmental Research Online (HERO) database5. Full details of the search strategy for each database are presented in the substance specific ETAP.
After deduplication in HERO9, records are imported into SWIFT Review10 software (Howard et al. 2016) to identify those references most likely to be applicable to a human health risk assessment. In brief, SWIFT Review has pre-set literature search strategies (“filters”) developed by information specialists that can be applied to identify studies that are more likely to be useful for identifying human health content from those that likely do not (e.g., analytical chemistry methods). The filters function like a typical search strategy: studies are tagged if the search terms appear in title, abstract, keyword, or medical subject headings (MeSH) fields. The applied SWIFT Review filters focus on lines of evidence: human, animal models for human health, and in vitro studies. The details of the search strategies that underlie the filters are available online11. Studies not retrieved using these filters are not considered further. Studies that include one or more of the search terms in the title, abstract, keyword, or MeSH fields are exported as a Research Information Systems (RIS) file for further screening in DistillerSR12, as described below.
3.2.2.3. Other Resources Consulted
The literature search strategies described above are designed to be broad; however, as with any search strategy, studies may be missed for assorted reasons (e.g., specific substance is not mentioned in title, abstract, or keyword content; inability to capture “grey” literature not indexed in the databases listed above). Thus, in addition to the database searches, the sources below are used to identify studies that may have been missed. Records that appear to meet the PECO criteria are uploaded into DistillerSR, annotated with respect to source of the record, and screened using the methods described in Section 3.2.3. Other sources consulted include:
- Manual review of the reference list from final or publicly available draft assessments [e.g., EPA Integrated Risk Information System (IRIS), Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological Profiles] or published journal review articles specifically focused on human health. Reviews may be identified from the database search or from ToxValDB.
- Manual review of the reference list of studies judged as PECO-relevant after full-text review.
- Electronic queries of European Chemicals Agency (ECHA) registration dossiers to identify data submitted by registrants13.
- Electronic queries of EPA ChemView database14 to identify unpublished studies, information submitted to EPA under Toxic Substances Control Act (TSCA) Section 4 (chemical testing results), Section 8(d) (health and safety studies), Section 8(e) (substantial risk of injury to health or the environment notices), and FYI (voluntary documents). Other databases accessible via ChemView include EPA’s High Production Volume (HPV) Challenge database and the Toxic Release Inventory (TRI) database.
- Electronic queries of NTP database of study results and research projects15.
- Manual review of the list of references in ECOTOX database for the substance(s) of interest18.
3.2.2.4. Confidential Business Information
The methods described above are intended to identify evidence that is in the public domain, but additional existing information may not be publicly available. To avoid mislabeling substances as lacking repeated dose toxicity studies, searches of Confidential Business Information (CBI) databases may also be conducted to confirm data availability status. Although the results of CBI studies cannot be considered in many assessment products (including IRIS, PPRTV, ATSDR), confirmation of a true lack of data is an important consideration when determining whether to initiate new toxicological studies. In certain cases, CBI information may be utilized to determine whether an ETAP should be developed.
3.2.3. SCREENING PROCESS
The studies identified from database searches and SWIFT Review are housed in the HERO system and imported into DistillerSR for title/abstract and full-text screening. Both title/abstract and full-text screening are conducted by two independent reviewers. Records that meet PECO criteria during title and abstract screening are considered for full-text screening. At both the DistillerSR title/abstract and full-text review levels, screening conflicts are resolved by discussion between the primary screeners with consultation by a third reviewer (if needed) to resolve any remaining disagreements. For citations with no abstract, the articles are initially screened based on all or some of the following: title relevance (title should indicate clear relevance) and length (articles two pages in length or less are assumed to be conference reports, editorials, or letters). During title/abstract or full-text level screening in DistillerSR, studies that did not meet the PECO criteria, but which could provide supporting information are categorized (or “tagged”) as supplemental information. Supplemental material is tagged using a “check all that apply” approach and reviewers resolve conflicts on the specific tags applied to studies.
Results of the screening process are presented in study flow diagrams and made publicly available in HERO to see full reference details. The study flow diagrams are also made available in an interactive literature tree format using EPA’s version of Health Assessment Workspace Collaborative (HAWC)19, a free and open-source web-based software application designed to manage and facilitate the process of conducting literature assessments.
3.2.4. EVALUATION OF WHETHER AVAILABLE STUDIES MAY PLAUSIBLY BE USED FOR POD AND REFERENCE VALUE DERIVATION
Studies that meet PECO criteria after full-text review are briefly summarized in DistillerSR. For animal studies, the following information is captured: chemical form, study type [i.e., acute (< 24 hours), short term (1-30 days), subchronic (30-90 days), chronic (>90 days20), reproductive, developmental], duration of exposure, route, species, strain, sex, dose or concentration levels tested, dose units, health outcome(s) and specific endpoint(s) assessed, and a summary of findings at the health outcome level [i.e., null or NO(A)EL/LO(A)EL based on author-reported statistical significance with an indication of which specific endpoints were affected].
For epidemiologic studies, the following information is summarized, when available: chemical form, population type (e.g., general population-adult, occupational, pregnant women, infants and children, etc.), study type (e.g., cross-sectional, cohort, case-control), short free text description of study population, sex, major route of exposure (if known), description of how exposure was assessed, health system and specific outcome assessed, and a summary of findings at the health system level (null or an indication of any associations found and a description of how the exposure was quantified in the analysis). Studies are extracted into DistillerSR or HAWC by one team member and checked by at least one other team member. These study summaries, referred to as a literature inventory, are presented in HAWC or Tableau visualization software,21 and are also available as an Excel file.
Studies in the literature inventory are analyzed with respect to suitability for the identification of an inhalation or oral POD, with preference given to the following:
- Animal studies with chronic or subchronic exposure durations.
- Animal study designs that assess effects of exposure on reproduction or development.
- Non-human mammalian studies using a species that is generally considered a relevant human surrogate.
- Animal studies with a broad exposure range and multiple exposure levels. These can provide information about the shape of the exposure-response relationship [see the EPA Benchmark Dose Technical Guidance, §2.1.1 EPA (2012b)] and facilitate extrapolation to more relevant (generally lower) exposures. However, single dose studies can be considered for reference value derivation if they test phenotypic health outcomes unexamined in multidose studies testing similar levels or for informing acute toxicity hazard(s).
- Human studies for which quantitative exposure measurements are available and exposure-response results are presented in sufficient detail (e.g., standardized mortality rate or relative risks, numbers of cases/controls). Studies based exclusively on duration of exposure analyses (i.e., longer versus shorter exposure duration) are typically not considered suitable for dose response unless additional information on exposure can be incorporated. Epidemiological studies that use biomarker measurements in tissues or bodily fluids as the metric for exposure are only considered suitable for dose response analysis if data or PBPK models are available to extrapolate between the reported biomarker measurement and the level of exposure.
- For both animal and human studies, the nature of the outcomes/endpoints assessed and whether these are interpretable with respect to potential adversity is considered. Typically, apical or clinical measures (“phenotypic”) are preferred over other endpoints for dose response. However, less direct endpoints (e.g., upstream precursors or biomarkers of exposure or effects known to precede an apical outcome) can be useful in dose response analyses when they can be reasonably established as predictive of, or strongly associated with, phenotypic outcomes interpreted as adverse.
3.3. EVIDENCE MAP REVIEW AND PRE-STUDY EVALUATION
The results of the evidence map are reviewed prior to initiation of the in vivo transcriptomic studies. Chemical substances may be eligible for an in vivo 5-day transcriptomic study and development of an ETAP if they meet one of the two following criteria: 1) confirmed to have no publicly available mammalian in vivo repeated dose toxicity studies or human studies suitable for POD and reference value derivation; or 2) the only available in vivo repeated dose studies have critical deficiencies and are considered uninformative using the study evaluation methods described by Thayer and colleagues (Thayer et al. 2022a; Thayer et al. 2022b). The EPA may also incorporate predictive methods (e.g., quantitative structure activity relationship (QSAR) models, analog approaches) to evaluate whether the standardized ETAP process is appropriate for a chemical substance or whether substantive modifications would be required.
3.4. 5-DAY IN VIVO TRANSCRIPTOMIC STUDIES AND ANALYSIS
A flow chart depicting the steps involved in the chemical procurement, analytical chemistry analysis, dose formulation and 5-day in vivo transcriptomic studies is provided in Figure 3-1.
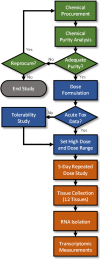
Figure 3-1
Flow chart depicting the main components and associated processes starting with chemical procurement and ending with the 5-day in vivo transcriptomic study. The green-colored processes and decision points are associated with the chemical procurement and (more...)
3.4.1. DOSE FORMULATIONS AND PRE-ADMINISTRATION ANALYSIS
3.4.1.1. Chemical Purity
Substances evaluated in an ETAP are typically procured from a commercial source, synthesized, or obtained from a reliable third party. The purity of the chemical substance is typically provided by the commercial source and also evaluated independently using the most appropriate analytical method [e.g., liquid chromatography-mass spectrometry (LCMS), gas chromatography-mass spectrometry (GCMS), Nuclear Magnetic Resonance (NMR)]. Quantitative structure activity relationship (QSAR) models may be used to identify the probable physical form, acidity, and analytical (Lowe et al. 2021; Mansouri et al. 2018; Mansouri et al. 2019)method. For most studies, the purity of single chemical test article of 95% or greater is acceptable. The purity of a single chemical test article less than 95% may be acceptable but will be documented accordingly. For mixtures, technical grade chemicals, and formulations, the relative purity and composition should reflect, as close as possible, the relevant human exposure context.
3.4.1.2. Vehicle Selection and Stability
For oral gavage studies22, a set of dosing vehicles are evaluated for chemical solubility and stability. The vehicles may include 1:1:8 Kolliphor:ethanol:deionized water, deionized water with ≤2% Tween® 80, corn oil, deionized water, as well as other options depending on physicochemical properties of the substance. The solubility is assessed visually and/or through analytical measurements. If an aqueous vehicle is used, the pH of the solution with test chemical should be determined, as too low or high of a pH can adversely affect the animal. Chemical stability in the dosing vehicle will also be assessed over a seven-day period.
3.4.1.3. Dose Identification
The approach used to select the dose range for the study will depend on a number of factors that may be specific to the substance of interest. For the ETAP study design, a minimum of eight dose levels plus a vehicle control will be evaluated. The dose range will be based on the highest dose with the dose levels decreasing at half-log10 intervals except for the lowest dose, which will be a full log10 lower than the second lowest dose. Given the intended application of ETAP and its pre-screening criteria, neither in vivo repeated dose toxicity data nor suitable human evidence will be available. Selection of the highest dose will depend on a number of factors that may be specific to the chemical of interest. If existing acute toxicity data are available for the substance of interest, the selection of the highest dose may consider the doses from such studies. If no acute toxicity data are available, in silico approaches (e.g., QSAR modeling) or pilot tolerability/dose range finding studies with limited numbers of animals may be used to inform selection of the highest dose.
3.4.2. ANIMAL HUSBANDRY AND EXPOSURE
Male and female Sprague Dawley (Crl:CD IGS, Charles River Laboratory) rats are purchased at 6 – 8 weeks of age. Upon receipt, the animals are placed on a standard, purified laboratory diet and reverse osmosis treated drinking water ad libitum. The specific brand and type of food and source of the water should be noted in Appendix II (Detailed Animal Study Report) of the individual assessment. After a 7- to 14-day quarantine and acclimation period, the animals are weighed and randomly assigned by weight to chemical exposure and control groups. Only clinically healthy animals are used in the study. The target age for initiating exposure is 8 - 10 weeks. For oral gavage studies, at least four male and four female rats per dose group receive the vehicle alone or test article in vehicle via gavage (5 or 10 ml/kg) for five days. Animals are weighed daily prior to administration and are observed twice daily, once during administration and once in the late afternoon, at least six hours apart, for assessment of moribundity and mortality. Formal clinical observations are performed on the first day post-dosing and prior to necropsy. Moribund animals or animals exhibiting overt clinical toxicity are removed from the study.
The temperature in the experimental animal room is maintained at a target of 22°C (± 3°C) with a relative humidity that is ideally between 50-60% but is at least 30% and preferably not to exceed 70% other than during room cleaning. Lighting is artificial with a sequence of 12 hours light, 12 hours dark. Animals are housed individually or caged in small groups of no more than three animals of the same sex in accordance with local institutional animal care and use requirements. The facility will be accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and will follow published Public Health Service animal care and use guidelines (NASEM 2011).
3.4.3. TISSUE COLLECTION
Optional blood samples may be collected at a specific time interval (e.g., 2 hr) following the first dose to provide estimates of toxicokinetic properties for certain chemicals. Treated and control animals are necropsied approximately 24 hours after the last exposure. Carbon dioxide asphyxiation is used as the method of euthanasia, with death confirmed by a secondary method such as exsanguination or cervical dislocation. At the time of necropsy, blood is collected [using potassium ethylenediaminetetraacetic acid (EDTA) as an anticoagulant] via cardiac puncture. Following collection, plasma is isolated and stored at approximately −80°C. While previous studies have demonstrated that transcriptional responses from the liver and kidney could be used as sentinels for phenotypic responses in other tissues (EPA 2024), a larger number of tissues will be dissected to increase the breadth of biological responses evaluated. The dissected tissues will include kidney, liver, adrenal gland, brain, heart, lung, ovary (females), spleen, testis (males), thyroid, thymus, and uterus (females). Tissue samples are typically collected within ten minutes of termination. The left liver lobe, right kidney, left lung, both testis, uterus, heart, spleen, thymus, and whole brain are sectioned into 5mm3 pieces. Samples from these larger tissues are then individually divided into at least three cryovials. At least two of the samples from each tissue in each animal are preserved in RNAlater™ (Thermo Fisher Scientific) at 4°C overnight and then frozen at approximately - 20°C for up to 3 weeks before transferring to approximately −80°C. At least one sample from each larger tissue is frozen immediately in liquid nitrogen and stored at approximately −80°C. The smaller bilateral tissues: adrenal glands, thyroid gland, and ovaries (female) are placed into two cryovials and preserved in RNALater with the left side of the tissue or gland going into the first cryovial and the right side going into the second cryovial. The first tube of RNALater preserved tissue is submitted for sequencing.
3.4.4. RNA ISOLATION AND TRANSCRIPTOMIC MEASUREMENTS
For each tissue undergoing transcriptomic analysis, total RNA is extracted from one of the aliquots stored in RNAlater™ using a standard approach for RNA isolation. RNA should be isolated and transcriptomic measurements performed on the kidney, liver, adrenal gland, brain, heart, lung, ovary (females), spleen, testis (males), thyroid, thymus, and uterus (females). The quantity and purity of the RNA (e.g., absorbance at 260 and 280 nm, absorbance at 260 and 230 nm, RNA integrity number) are determined and documented. The isolated total RNA is used to perform targeted RNA sequencing (RNA-seq) using the BioSpyder TempO-Seq rat S1500+ assay according to manufacturer’s instructions. No specific RNA purity or integrity criteria are applied to the RNA samples as the TempO-Seq assay has been designed to provide high quality gene expression measurements on whole cell lysates, purified RNA, and formalin-fixed paraffin embedded tissue. Each sample is sequenced to a target read depth of at least 1 million mapped reads per sample.
3.4.5. TRANSCRIPTOMIC DATA ANALYSIS
A flow chart depicting the steps involved in the transcriptomic data processing, dose response analysis, gene set summarization, and derivation of the TRV is provided in Figure 3-2.
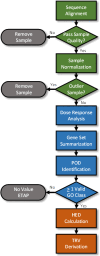
Figure 3-2
Flow chart depicting the main components and associated processes in the transcriptomic data analysis and TRV derivation. The green-colored processes and decision points are associated with the sequence processing, normalization, and quality evaluation. (more...)
3.4.5.1. Sequence Alignment
Raw sequencing reads (FASTQ files) are aligned to known probe sequences listed in the TempO-Seq probe manifest to compute a matrix of read counts for each probe in each sample. Initial quality checks are performed post-alignment to identify samples with insufficient sequencing depth or input RNA to yield reliable results. Each FASTQ file is aligned to the TempO-Seq probe manifest using HISAT2 (Kim et al. 2015; Kim et al. 2019). The alignment results are imported directly into SAMtools (Li et al. 2009) to compute probe-level counts for each individual FASTQ file. Samples are examined for additional quality statistics and those not meeting minimum quality standards are removed from the analysis. Samples that do not pass quality checks may be subjected to reprocessing for RNA isolation and RNA-seq. Quality metrics include (Harrill et al. 2021a):
- Sequencing depth (i.e., total number of mapped reads). Samples with < 10% of target depth are removed from further analysis.
- Fraction of uniquely mapped reads. Samples with < 50% of reads uniquely mapped to known probes are removed from further analysis.
- Probe coverage (i.e., total probes with at least 5 reads). Samples with < 1,200 covered probes are removed from further analysis.
- Signal distribution (i.e., the minimum number of probes that capture 80% of total mapped reads in the sample). No cutoff is applied, but this metric is considered when evaluating potential outlier samples (see below).
3.4.5.2. Sample Normalization
Prior to performing downstream gene expression across samples, probe counts for each sample are normalized to adjust for differences in sequencing depth. For each exposure regimen in each sex and tissue, raw probe counts for all samples (including matched controls) are normalized within each sample as follows:
- All probes with a mean read count < 5 are removed, as these probes lack sufficient signal for reliable analysis.
- Each remaining probe is normalized to Counts Per Million (CPM), which is probe count * 1,000,000 / sum of all remaining probe counts in sample.
- CPM values are transformed to log2 scale with added pseudo-count of 1 to prevent taking log of zero counts and ensuring a positive value for dose response modeling.
To identify potential outlier samples or batch effects, a principal component analysis (PCA) is performed on subsets of samples corresponding to: 1) all samples corresponding to same substance, tissue, and sex, including matched vehicle controls (“chemical exposure PCA”); and 2) all matched vehicle controls corresponding to the same tissue and sex (“vehicle PCA”). Samples not meeting the sequencing quality metrics (e.g., < 50% of uniquely aligned reads) are excluded prior to PCA analysis. Outlier samples are identified based on the following considerations:
- Individual samples separated from all remaining samples on either principal component #1 (PC1) or principal component #2 (PC2) by >2x the span of all other samples on the corresponding PC are considered strong outliers and removed from further analysis.
- Individual samples separated by <2x the range of all other samples are considered moderate outliers, and additional exclusion criteria are considered:
- Vehicle samples that appear as moderate outliers on both a chemical exposure PCA and vehicle PCA are excluded unless multiple controls from the same group appeared as outliers.
- Moderate outlier samples with lower quality than corresponding tissue samples by one or more sequencing quality metrics (e.g., percentage of uniquely mapped reads) are excluded.
- Samples that appear as moderate outliers in both PC1 and PC2 with a relatively large Euclidean distance from all other remaining samples are excluded.
- Moderate outlier samples that are especially distant from corresponding replicates or similar doses are excluded.
When multiple outlier samples are present on the same PCA, they are only removed if each outlier sample corresponds to a different dose group, as these are unlikely to represent any reproducible dose-dependent effect. A minimum number of two samples that pass quality control and outlier detection is required for each dose level; individual dose levels not meeting this criterion will be excluded from subsequent analysis. A minimum number of three vehicle control samples that pass quality control and outlier detection is required to proceed with dose response modeling for a given tissue, sex, and exposure regimen.
3.4.5.3. Dose Response Analysis
Once the sequencing data are aligned and normalized, and all low quality and outlier samples removed, dose response modeling is performed. Each data set consists of the series of remaining replicates for all concentrations of a single chemical and matched vehicle controls in the same sex and tissue. The dose response modeling is performed independently on each probe and for each data set using the peer-reviewed BMDExpress software version 2.3 (Phillips et al. 2019; Yang et al. 2007). The dose response analysis procedures are consistent with the NTP Approach to Genomic Dose Response Modeling (NTP 2018), but have been adapted for the specific gene expression platform used in this method (EPA 2024):
- Normalized Log2(CPM) with added pseudo-count of 1 is used as input.
- For each data set (specific combination of exposure, sex, and tissue), the analysis of variance (ANOVA) pre-modeling test is used to confirm that at least one probe has significant response with a false discovery rate (FDR) < 0.05. If no probes have a significant response, the particular sex and tissue combination is determined to be inactive for the chemical and dose range tested.
- Pre-filtering of probes suitable for dose response modeling is performed using a William’s trend test (p < 0.05) and a mean absolute fold-change relative to vehicle controls of 1.5x or greater in at least one dose.
- Model fitting and BMD determination are performed on each probe passing the pre-filtering criteria:
- The following dose response models are used in the analysis – linear, second degree polynomial, power, Hill, second degree exponential, third degree exponential, fourth degree exponential, and fifth degree exponential.
- Models are run assuming a constant variance.
- For the power model, power is restricted >=1.
- The model with the lowest Akaike information criterion (AIC) is selected as the best-fit model except in cases where the “k” parameter for the Hill model is less than one-third the lowest dose. In cases for which the “k” parameter for the Hill model is out of bounds, the Hill model is excluded from the final selection (Rowlands et al. 2013; Thomas et al. 2013b).
- The Benchmark Response (BMR) is set to 1.349 * standard deviation of replicate vehicle control samples (Thomas et al. 2007). Based on EPA guidance, a BMR of 1 standard deviation for continuous data approximates a 10% increase in risk for normally distributed effects when the direction of the effects is known (EPA 2012). However, for most gene expression changes, the direction is not known a priori. To provide an equivalent 10% increase in risk, a BMR of 1.349 * standard deviation is required (Thomas et al. 2007).
- The BMD, BMDL, and BMD upper confidence bound (BMDU) are calculated for each probe.
3.4.5.4. Gene Set Summarization
BMD results for each exposure/sex/tissue are aggregated into Gene Ontology (GO)23 biological process classes to identify BMD values. The gene set summarization process is performed as follows:
- Probes are mapped to associated genes. For genes with multiple probes, the BMD/BMDL values from valid probes are averaged. Probes mapping to multiple genes are excluded.
- Genes with conflicting probes are flagged for further review. Using the default setting in BMDExpress, conflicting probes are defined as those with a correlation cut-off of < 0.5 across doses.
- The BMD values for the individual genes are aggregated into GO biological process classes using the current annotations available in BMDExpress.
- GO classes containing fewer than 3 genes with valid BMDs meeting the above criteria are removed from the analysis.
- The BMD and BMDL for each GO class are calculated as the respective medians of corresponding values from the associated genes.
3.4.5.5. POD Identification
The GO biological process class with the lowest median BMD value is identified separately for each tissue examined in each sex. For each tissue and sex, if the lowest median BMD corresponds to more than one GO biological process class, the GO class with the lowest median BMDL value is selected. If there is more than one GO biological process class identified with identical median BMD and BMDL values for the same tissue and sex, then the following criteria are used in the order provided to select the most informative GO class:
- Only the GO classes with the highest number of dose-responsive genes are retained.
- Only the GO classes with the highest percent coverage of the gene set are retained.
- Only the GO classes with the highest (most specific) GO level are retained.
If multiple GO classes remain after applying the selection criteria above, then the additional remaining GO classes will be reported in a footnote.
If the lowest median BMD value identified across all tissues and each sex is more than 3-fold below the lowest positive dose, a ‘no value’ ETAP is declared, and the dose range tested is reported. A follow-up study with an extended dose range may be considered. If the lowest median BMD value identified is less than 3-fold below the lowest positive dose or within the tested dose range, the ETAP is considered valid. The GO biological process class with the lowest median BMD value across all tissues and each sex is then identified. If the lowest median BMD is identical in more than one tissue or sex, the GO biological process class with the lowest median BMDL value is selected. If there is more than one tissue or sex with identical median BMD and BMDL values, then the same criteria described above are used to select the most informative GO biological process class. The median BMDL associated with the identified GO biological process class is selected as the transcriptomic POD. The transcriptomic POD is defined as the dose at which there were no coordinated transcriptional changes that would indicate a potential toxicity of concern. The coordinated transcriptional changes used to identify the POD do not necessarily discriminate between specific hazards, adverse or adaptive effects, nor are they used to infer a mechanism or mode of action. If no tissue in either sex passes the pre-modeling filter nor produces at least one valid GO class, a ‘no value’ ETAP is declared and the dose range tested is reported. A follow-up study with an extended dose range may be considered.
3.5. HUMAN EQUIVALENT DOSE
The selected transcriptomic BMDL is scaled to a Human Equivalent Dose (HED) using an oral dosimetric adjustment factor (DAF) based on interspecies body weight allometry (EPA 2011a)(Fig. 3-2). The BMDLHED is calculated using the following equation:
The BWRat is the study-specific mean terminal rat body weight for the sex that is associated with the POD. The BWHuman is the reference human body weight of 80 kg (EPA 2011b). The BMDLHED represents the POD used to derive the TRV. The BMDLHED is also provided in the ETAP to enable users to calculate values for varying risk assessment applications such as a margin of exposure (EPA 2000, 2012), and to evaluate potential health risks from chemical mixtures (EPA 2000). Context specific applications are dependent upon multiple factors, including the statute or legislative mandate/purview involved, the exposure situation being addressed, the hazard and dose response data available and associated uncertainties, and the fit-for-purpose needs of the decision-maker.
3.6. TRANSCRIPTOMIC REFERENCE VALUES
Biological process-based, transcriptomic PODs obtained from the 5-day in vivo oral exposure studies, described in Sections 3.4 and 3.5 of this document (Figs. 3-1 and 3-2), may be used in the derivation of TRVs through application of uncertainty factors (UFs). The UFs are consistent with traditional human health assessment guidance and the fit-for-purpose rationale(s) considered for quantitative application of each factor are provided below24.
3.6.1. UNCERTAINTY FACTORS
As a common practice in human health risk assessment of oral exposures, UFs are used in deriving reference dose (RfD) values from PODs estimated using experimental data (EPA 1994, 2002). UFs are intended to account for: 1) unknown or imprecise measures of variability in sensitivity among the members of the exposed human population (i.e., interhuman or intraspecies variability, UFH); 2) the uncertainty in extrapolating animal data to humans (i.e., interspecies variability, UFA); 3) the uncertainty in extrapolating from data obtained in a study with less-than-lifetime exposure to lifetime exposure (e.g., extrapolating from subchronic to chronic exposure, UFS); 4) the uncertainty in extrapolating from a lowest observed adverse effect level (LOAEL) rather than from a no observed adverse effect level (NOAEL) (UFL); and 5) the uncertainty associated with deficiencies or knowledge gaps in the chemical-specific database (UFD).
In current EPA human health risk assessment practice, in the absence of chemical-specific data supporting quantitative application of uncertainty, standard UFs of 10 are recommended, with 3 used in place of half-power values (i.e., 100.5) if some aspect of uncertainty is accounted for, or if uncertainty is not comprehensively addressed. A UF of 1 is applied if either the uncertainty is not relevant (e.g., UFL of 1 because the POD is a BMD value), or if qualitative evidence comprehensively characterizes an area of uncertainty. Within the scope of an ETAP, the initial step in the process for selecting and pre-qualifying chemicals occurs through systematic evidence mapping to ensure that only substances with no existing or publicly accessible repeated dose toxicity studies or human evidence suitable for use as a POD and reference value derivation are considered. In the rare case that information is surfaced for the target chemical that informs some aspect of a given area of uncertainty, there may be an opportunity to adjust quantitative uncertainty application(s). Scientific support for application of UFs to PODs in the derivation of reference values should be clearly documented, with the qualitative and quantitative rationale defined explicitly.
3.6.1.1. Intraspecies Variability Uncertainty Factor (UFH)
The intraspecies UFH is applied to account for variation in susceptibility within the human population (interindividual variability) and the possibility (given a lack of relevant data) that the database available is not representative of the exposure/dose response relationship in the subgroups of the human population that are most sensitive to the health hazards of the substance being assessed. As the reference dose is defined to be applicable to “susceptible subgroups,” this UF is used to account for uncertainty in that regard. The adjustment of the intraspecies UFH from 10 should be considered only if data are sufficiently representative of the exposure/dose response data for the most susceptible human population(s) (e.g., early and late lifestages). The UFH may be presumed to entail aspects of both toxicokinetic (TK) and toxicodynamic (TD), thus providing an opportunity to integrate traditional and/or NAM-based information that might support reduction in the UF or quantitative application of a data-derived extrapolation factor (DDEF) for human TK (DDEFHK) and/or human TD (DDEFHD) for the UFH (EPA 2014).
For transcriptomic PODs identified in the ETAP, a UFH of 10 is applied. However, if information is available that informs intraspecies variability or unique sensitivities or susceptibilities of relevance to human populations (e.g., toxicokinetic and/or toxicodynamic variation[s] in human populations), then expert judgment may be used to consider the weight of the evidence to support application of a DDEFHK and/or DDEFHD in place of the standard UFH of 10. However, should human intraspecies information be identified during the evidence mapping phase, consideration should be given to transitioning such a substance to another assessment product line outside of ETAP.
3.6.1.2. Animal-to-Human Interspecies Uncertainty Factor (UFA)
The interspecies UFA is applied to account for the extrapolation of laboratory animal data to humans, and it generally is presumed to include cross-species TK and TD uncertainties. With chemical-specific data that informs cross-species scaling of TK (e.g., clearance or plasma T1/2), the TK half of the UFA may be reduced from a 3 (i.e., 100.5) to a 1 through the development and application of a dosimetric adjustment factor (DAF) that accounts, in general, for differences in TK between animals and humans. In the absence of chemical-specific TK data, a DAF may be applied to a transcriptomic POD obtained from in vivo animal oral exposure study designs using standard EPA guidance and practice, such as BW3/4 allometric scaling (EPA 2011a). This results in the derivation of a POD human equivalent dose (PODHED, such as a transcriptomic BMDLHED).
The UFA is intended to also account for differences in TD-related species sensitivity between the laboratory animals used for testing and humans. Seldom are there chemical-specific data available to inform TD differences between species, and one-half the standard 10-fold interspecies UFA (i.e., 100.5) is assumed to account for such differences. Unless data support the conclusion that the laboratory test species is more or equally as susceptible to a chemical substance as are humans, and in the absence of any other specific TK or TD data, a UFA of 3 (in conjunction with calculation of a PODHED) is applied for the ETAP.
3.6.1.3. Subchronic-to-Chronic Duration Uncertainty Factor (UFS)
EPA defines a chronic duration as repeated exposure by the oral, dermal, or inhalation route for more than approximately 10% of the life span in humans, corresponding to more than approximately 90 days to 2 years in typically used laboratory animal species (EPA 2002, 2011a). Subchronic duration is defined as repeated exposure by the oral, dermal, or inhalation route for more than 30 days, up to approximately 10% of the life span in humans (more than 30 days up to approximately 90 days in traditional laboratory animal species) (EPA 2002, 2011a). In traditional risk assessment practice, if no chronic duration study is available, information from a subchronic study may be used to support the derivation of an RfD with the application of a UFS of 10 to the subchronic POD.
Duration extrapolation in the context of an ETAP is informed by multiple previous studies that have demonstrated dose-concordance between traditional apical effect-based PODs derived from longer-term (i.e., chronic) duration studies and gene set-based transcriptomic PODs derived from shorter-term studies (EPA 2024). The concordance was robust across species, sexes, routes or modes of exposure, and technological platforms. In the analysis performed to inform the choices and parameters used in the transcriptomic dose response modeling process, the error in the concordance of the 5-day transcriptomic BMDs with the apical effect BMDs from chronic rodent bioassays was approximately equivalent to the combined inter-study variability associated with the 5-day transcriptomic study and the chronic rodent bioassay (EPA 2024). This demonstrates that the observed differences between the 5-day transcriptomic and chronic apical BMDs are largely driven by inter-study variability in the BMDs, rather than systematic differences. As a result, when using 5-day transcriptomic PODs for non-cancer health effect domains in the ETAP, a UFS of 1 is applied for considerations of duration in the derivation of a TRV.
3.6.1.4. Lowest Observed Adverse Effect Level (LOAEL)-to-No Observed Adverse Effect Level (NOAEL) Uncertainty Factor (UFL)
The current EPA approach for dose response assessment prioritizes the application of BMD modeling to identify potential PODs for effects. However, in traditional human health risk assessment practice, when dose response data are not amenable to BMD modeling, point estimates such as LOAELs and NOAELs are identified as potential PODs. A LOAEL is defined as the lowest exposure level at which there are statistically and/or biologically significant increases in frequency or severity of adverse effects between an exposed population and a corresponding control group. A NOAEL is the highest dose level tested at which the specified adverse effect is not produced. Generally, a LOAEL-to-NOAEL uncertainty factor (UFL) is applied to derive a non-cancer reference value using an apical effect LOAEL if a NOAEL is unavailable. This UFL is employed to estimate an exposure level below the LOAEL expected to be in the range of a NOAEL. Importantly, the underlying biology leading to and/or resultant of cell, tissue, or organ/system level toxicity invariably involves changes in gene expression. Selecting the gene set with the lowest BMD and BMDL is not necessarily associating transcriptional events with a specific adverse event per se, rather, it is thought to be a dose that approximates a NOAEL.
The gene set summarization of the gene expression changes is described in Section 3.4.5.4 and is suggested as the minimum unit of transcriptional activity to be used in the identification of a POD. That is, BMDLs for single genes are not recommended for POD identification; rather, only those groupings of genes that constitute a GO biological process class in accordance with the criteria outlined in 3.4.5.4 are considered for potential POD (e.g., GO biological process-based BMDL) identification. When GO biological process-based BMDL values are successfully identified for one or more classes using methods consistent with the ETAP, a UFL of 1 is applied.
3.6.1.5. Database Uncertainty Factor (UFD)
In traditional human health risk assessment, the UFD is intended to account for the potential for deriving an under-protective RfD as a result of an incomplete characterization of the substance’s toxicity via the oral exposure route. In addition to identifying data gaps in toxicity information, review of existent data may also suggest that a lower reference value might result if additional data are available. Consequently, in deciding to apply this factor to account for deficiencies in the available data set and in identifying its magnitude, the assessor should consider both the data lacking and the data available for health outcome domains, tissues, or organ systems, as well as life stages. In the context of the ETAP, previous studies have demonstrated that GO biological process-based transcriptomic BMD values following 5 days of exposure are in agreement with BMD values for histopathological effects in two-year chronic rodent bioassays (EPA 2024). Responses in other health effect domains, such as developmental, reproductive, endocrine, neurotoxicity, or immunotoxicity, may not necessarily be accounted for in 5-day in vivo transcriptomic studies. Therefore, a UFD of 10 should be applied to account for data gaps in the derivation of a TRV for an ETAP.
3.6.1.6. Derivation of the Transcriptomic Reference Value
Using the BMDLHED from Section 3.5, the standard calculation of the TRV is summarized based on the following equation; however, the exact calculation may vary in unusual circumstances based on the considerations discussed above:
The TRV is defined as an estimate of a daily oral dose to the human population that is likely to be without appreciable risk of adverse non-cancer health effects over a lifetime. The TRV is derived from a transcriptomic POD with uncertainty factors applied to reflect limitations of the data used. While a TRV is expressly presented as a chronic value in an ETAP, it may also be applicable across other exposure durations of interest including short-term and subchronic. This approach has been previously used by EPA in certain risk assessment applications (e.g., PPRTV assessments) wherein a chronic non-cancer reference value has been adopted as a conservative estimate for a subchronic non-cancer reference value when data quality and/or lack of duration relevant hazard and dose response data preclude direct derivation.
3.7. ETAP REPORTING
The summary results from the systematic evidence mapping, 5-day in vivo transcriptomic study, and TRV are to be reported in a standardized ETAP reporting template (Appendix). The use of the Organization for Economic Cooperation and Development’s (OECD) Omics Reporting Template (Harrill et al. 2021b; OECD 2022) as an appendix to the ETAP reporting template is recommended.
3.8. INTERNAL AND EXTERNAL REVIEW OF ETAPs
The methods for developing the ETAP outlined in this document have been internally reviewed by ORD scientists and management. The methods have also been externally peer-reviewed by the EPA Board of Scientific Counselors and subject to public comment.
All ETAP activities and testing are covered under a standard EPA Category A Quality Assurance Project Plan (QAPP). Each ETAP will undergo an Audit of Data Quality (ADQ) by an EPA Quality Assurance (QA) team. For ETAPs that follow the standardized methods, the individual assessment will undergo internal review by at least two ORD technical experts but will not receive independent external peer review. The EPA BOSC has endorsed not adding external peer review for individual ETAPs that are the product of a peer reviewed and approved standardized process without assessment or judgments. For ETAPs that have substantive modifications to the standardized methods, the individual assessment will undergo internal review by at least two ORD technical experts, and independent external peer review. Examples of substantive modifications may include application of a DDEF, change in a standardized UF, or change in the DAF. The EPA BOSC has endorsed adding a limited scope external peer review if the EPA determines it is necessary to depart from the standard process. All ETAPs will be published on a publicly available EPA ORD website (https://www.epa.gov/etap).
Footnotes
- 3
ToxValDB is a database designed to store a wide range of public toxicity information while maintaining the linkages to original source information so that users can access available details. ToxValDB collates publicly available toxicity dose–effect related summary values typically used in risk assessments. These include POD data collected from data sources within ACToR and ToxRefDB, and no-observed and lowest-observed (adverse) effect levels (NOEL, NOAEL, LOEL, LOAEL) data extracted from repeated dose toxicity studies submitted under REACH (Regulation for Registration, Evaluation, Authorisation and restriction of chemicals in the EU). Also included are reference dose and concentration values (RfDs and RfCs) from EPA’s IRIS and Provisional Peer-Reviewed Toxicity Values (PPRTV) assessments. Acute toxicity information is extracted from a number of different sources, including OECD eChemPortal, ECHA, NLM HSDB (Hazardous Substances Data Bank), ChemIDplus via EPA TEST (Toxicity Estimation Software Tool), and the EU JRC (Joint Research Centre) AcutoxBase. Finally, data from the eChemPortal and the EU COSMOS project also are included in ToxValDB. The ToxVal database is available through the EPA CompTox Chemicals Dashboard at: https://comptox
.epa.gov/dashboard. - 4
The EPA CompTox Chemicals Dashboard is available at: https://comptox
.epa.gov/dashboard/ - 5
EPA’s HERO database provides access to the scientific literature behind EPA science assessments. The database includes more than 3 million scientific references and associated data from the peer-reviewed literature used by EPA to develop reports that support critical agency decision making and regulations.
- 6
The PubMed database is available at: https://pubmed
.ncbi.nlm.nih.gov/ - 7
The Web of Science database is available at: https://www
.webofscience.com/ - 8
The ProQuest database is available at: https://www
.proquest.com/ - 9
Deduplication in HERO involves first determining whether a matching unique ID exists (e.g., PMID, WOSid, or DOI). If one matches one that already exists in HERO, HERO will tag the existing reference instead of adding the reference again. Second, HERO checks if the same journal, volume, issue and page number are already in HERO. Third, HERO matches on the title, year, and first author. Title comparisons ignore punctuation and case.
- 10
SWIFT-Review is an interactive workbench of tools to assist with problem formulation and literature prioritization. SWIFT is an acronym for Sciome Workbench for Interactive computer-Facilitated Text-mining. The workbench is available at: https://www
.sciome.com/swift-review/ - 11
Swift-Review filters are available at: https://www
.sciome.com /swift-review/searchstrategies/ - 12
DistillerSR is a web-based systematic review software used to screen studies available at: https://www
.evidencepartners .com/products /distillersr-systematic-review-software. - 13
ECHA registration dossiers available at: https://echa
.europa.eu /information-on-chemicals /information-from-existing-substances-regulation - 14
EPA ChemView database is available at: https://chemview
.epa.gov/chemview/ - 15
NTP data and resources are available at: https://ntp
.niehs.nih .gov/data/index.html - 16
OECD Existing Chemicals Database is available at: https:
//hpvchemicals .oecd.org/ui/Default.aspx - 17
OECD eChem Portal is available at: https://www
.echemportal .org/echemportal/substance-search - 18
EPA’s ECOTOX Knowledgebase is available at: https://cfpub
.epa.gov/ecotox/ - 19
EPA’s Health Assessment Workspace Collaborative (HAWC) is available at: https://hawc
.epa.gov - 20
EPA considers chronic exposure to be more than approximately 10% of the life span in humans. For typical laboratory animal species, this can lead to consideration of exposure durations of approximately 90 days to 2 years. However, studies in duration of 1 - 2 years are typical of what is considered representative of chronic exposure rather than durations just over 90 days.
- 21
Tableau is available at: https://www
.tableau.com - 22
Current application is limited to oral gavage studies. Certain toxicological responses are route and dosing regimen specific. As a result, other routes of exposure may be considered in the future. Extrapolations to other routes and dosing regimens may potentially be considered for gap-filling under specific circumstances.
- 23
Additional information on the Gene Ontology (GO) knowledgebase may be accessed at: http://geneontology
.org/ - 24
The scientific basis underlying selection of the default uncertainty factors for the ETAP will be periodically reviewed. If adjustments are needed and justified, the revised ETAP Standard Methods will undergo external peer review as appropriate and consistent with EPA ORD processes.
Publication Details
Copyright
Publisher
U.S. Environmental Protection Agency, Washington (DC)
NLM Citation
Auerbach S, Casey W, Chang D, et al. Standard Methods for Development of EPA Transcriptomic Assessment Products (ETAPs). Washington (DC): U.S. Environmental Protection Agency; 2024 Mar. 3, METHODS.

