Congenital Insensitivity to Pain Overview
Katherine Rose Schon, MB ChB, MRCP, MA, Alasdair Patrick John Parker, MBBS, MRCP, MD, MA, and Christopher Geoffrey Woods, MB ChB, FRCP, FMedSci.
Author Information and AffiliationsInitial Posting: February 8, 2018; Last Revision: June 11, 2020.
Estimated reading time: 24 minutes
Summary
The goals of this overview on congenital insensitivity to pain (CIP) are the following.
Goal 2.
Review the causes of congenital insensitivity to pain.
Goal 3.
Provide an evaluation strategy to identify the genetic cause of congenital insensitivity to pain in a proband.
Goal 4.
Provide a brief summary of management of congenital insensitivity to pain.
1. Clinical Characteristics of Congenital Insensitivity to Pain
Congenital insensitivity to pain (CIP) is an extremely rare phenotype characterized by the inability to perceive pain (absence of nociception) from birth. Individuals with CIP do not feel pain from any noxious stimuli, including inflammation and heat [Goldberg et al 2007]. This review does not cover conditions that cause a generalized sensory neuropathy.
Inability to feel pain leads to repeated injuries and prevents normal healing.
Characteristic Findings
Eyes
All affected individuals are at risk for corneal injuries due to absent corneal reflexes.
Permanent corneal scarring can develop and is best assessed through a slit-lamp examination. It has been observed clinically that corneal transplants have an increased risk of failure, presumably due to the lack of nociceptive innervation of the new cornea and the continuing lack of nociceptive innervation of the conjunctiva.
Emotional tearing, as opposed to pain induced tearing, is likely to be present [
Shatzky et al 2000].
Infections
Apparent selectively reduced immunity to Staphylococcus aureus has been observed in some affected individuals, leading to recurrent soft tissue infections, abscesses, and osteomyelitis.
Temperature Regulation, Anhidrosis, and Hyperhidrosis
Some individuals have anhidrosis (lack of sweating), which disturbs thermoregulation and can lead to recurrent episodes of unexplained fever [Indo et al 1996, Indo 2001] (see Table 1).
Marked hyperhidrosis may be seen in those affected individuals who have a heterozygous pathogenic c.2432T>C (p.Leu811Pro) variant in SCN11A [Woods et al 2015].
Hyperpyrexia can be fatal if untreated [Shatzky et al 2000].
Hypothermia can occur in cold conditions.
Development and Intellect
Development and intellect may be normal or delayed/disabled (see Table 1).
Other
Chronic anemia of unknown cause was observed in 22/28 Israeli affected individuals with congenital insensitivity to pain and anhidrosis [
Shatzky et al 2000].
A few individuals have neuropathic pain, although this does not limit activities [
Wheeler et al 2014; Author, personal observation].
Establishing the Clinical Diagnosis of Congenital Insensitivity to Pain
There are no consensus clinical diagnostic criteria for CIP. However, a diagnosis requires visible proof of lack of nociception in a conscious individual of normal intellectual ability. In those with intellectual disability CIP may be more difficult to diagnose clinically.
Clinical Examination
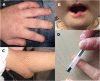
Nociception is assessed by applying painful stimuli, which in people with normal nociception would be so difficult to bear that they would move the part of the body away from the stimulus and/or express discomfort. The authors have experience of children being incorrectly judged to have insensitivity to pain after an inadequately painful stimulus.
The technique should not damage/scar prior to significant pain (e.g., sternal rub, which bruises before significant pain).
Application of 5-10 kg pressure with a pen pressed onto the nail bed (the nail bed blanches for a few seconds afterward) is reliable (see ).
Assessment of the remainder of the peripheral and central nervous system is typically normal (touch, vibration and position sense, motor functions, and deep tendon reflexes).
Supportive Laboratory Findings
Routine nerve conduction studies and electromyogram are typically normal [Shatzky et al 2000].
For more information about autonomic function testing for CIP with anhidrosis, click here (pdf).
Nerve biopsy is not routinely performed in clinical practice. Skin biopsy to determine intra-epidermal nerve fiber density and autonomic innervation is performed in adults in some centers.
2. Causes of Congenital Insensitivity to Pain
All causes of CIP affect nociceptors (specialized peripheral sensory neurons) and either cause nonfunctional nociceptors or failure of nociceptor neurodevelopment [Nahorski et al 2015b]. Congenital insensitivity to pain is an extremely rare phenotype and the exact proportion of individuals with pathogenic variants in each gene within the whole population is not known.
Table 1.
Genes Associated with Congenital Insensitivity to Pain (CIP)
View in own window
| Gene | Proportion of Affected Individuals with Mutation of This Gene | MOI | Distinguishing Features |
|---|
| CLTCL1 1 | Rare | AR |
|
| NGF 3 | Rare | AR | Variable phenotype Individuals w/biallelic null variants may have anhidrosis, mild/moderate ID, prematurely aged appearance, ↑ Staphylococcus aureus infections, & Charcot joints. Individuals w/a homozygous missense variant had impairment of pain/temperature sensation & Charcot joints, normal intellect & normal sweating. 4
|
| NTRK1 5 | Common | AR | Anhidrosis Tendency to develop corneal ulcers that heal poorly 6 ID in a majority; always less intellectually able than unaffected family members Predisposition to Staphylococcus aureus infections Charcot joints Dry skin w/lichenification
|
| PRDM12 7 | Intermediate | AR | Non-global pain insensitivity in some Absent corneal reflex & impaired tear production Staphylococcus aureus infections No Charcot joints Difficulties w/temperature regulation in some Usually normal neurologic exam, development, intellect, & olfaction 8 Known as HSAN VIII
|
| SCN9A
9, 10 | Common | AR |
|
| SCN11A 12 | Rare | AD | Delayed motor development Mild muscle weakness Joint hypermobility Gastrointestinal dysfunction (intestinal hypoperistalsis or diarrhea) Pruritis Hyperhidrosis in those w/c.2432T>C (p.Leu811Pro) variant
|
| ZFHX2 13 | Rare | AD | 1 family reported Non-global pain insensitivity w/lower back pain, headaches, & pain during childbirth perceived Normal intelligence Scarce or absent sweating Variably reduced sensitivity to heat and cold Low sensitivity to capsaicin – able to eat large amount of hot pepper w/out discomfort Some autonomic features such as fainting & vomiting
|
AD = autosomal dominant; AR = autosomal recessive; HSAN = hereditary sensory and autonomic neuropathy; ID = intellectual disability; MOI = mode of inheritance
- 1.
- 3.
- 4.
Three individuals from a large northern Swedish family who were homozygous for the NM_002506.2:c.661C>T, (p.Arg211Trp) pathogenic variant [Einarsdottir et al 2004]. A proportion of adults who were heterozygous for the NM_002506.2:c.661C>T, (p.Arg211Trp) pathogenic variant in this family had mild or moderate problems with joint deformities but were not believed to actually be affected by CIP.
- 5.
- 6.
- 7.
- 8.
Saini et al [2017] described a male age two years with a homozygous pathogenic splice site variant in PRDM12 who had global developmental delay, dolicocephaly, frontal bossing, and deep-set eyes as well as congenital insensitivity to pain.
- 8.
- 10.
Pathogenic variants are typically truncating, although one missense variant and one in-frame deletion have been described [Cox et al 2010].
- 11.
- 12.
- 13.
Table 2.
Other Disorders to Consider in the Differential Diagnosis of Congenital Insensitivity to Pain (CIP)
View in own window
| Differential Diagnosis Disorder | Gene(s) | MOI | Clinical Features of the Disorder |
|---|
| Overlapping w/CIP | Distinguishing from CIP |
|---|
|
Hereditary
|
|
Hypohidrotic ectodermal dysplasia
|
EDA
EDAR
EDARADD
| XL
AR
AD |
| Insensitivity to pain not a feature |
|
Lesch-Nyhan syndrome
|
HPRT1
| XL | Progressive self-injurious behavior (biting fingers, hands, lips, cheeks; banging the head or limbs) |
|
|
COL1A1/2-related osteogenesis imperfecta
|
COL1A1
COL1A2
| AD | Multiple fractures | Fractures cause pain Fractures occur w/minimal or absent trauma Assoc w/other features incl blue sclera, short stature, joint hypermobility, deafness
|
| Familial dysautonomia (also known as HSAN III) | ELP1 (IKBKAP) | AR | Reduced pain from birth | Gastrointestinal dysfunction, vomiting crises, recurrent pneumonia, cardiovascular & temperature instability |
|
MPV17-related hepatocerebral mitochondrial DNA depletion syndrome
|
MPV17
| AR |
| Infantile-onset liver dysfunction typically → liver failure Failure to thrive, lactic acidosis, & hypoglycemia More severe neurologic involvement; may incl white matter abnormalities on MRI & seizures
|
|
Acquired
|
| Leprosy 1 | NA | NA | Insensitivity to pain Painless injuries
| Skin lesions (hypopigmented macules, nodules, plaques, or diffuse skin infiltration) Enlargement of peripheral nerves Localized (not universal) insensitivity to pain
|
| Non-accidental / abusive injury | NA | NA | Multiple unexplained injuries |
|
AD = autosomal dominant; AR = autosomal recessive; DD = developmental delay; HSAN = hereditary sensory and autonomic neuropathy; ID = intellectual disability; MOI = mode of inheritance; NA = not applicable; XL = X-linked
- 1.
Prevalence
The prevalence of the CIP phenotype is unknown. Consideration of this diagnosis has increased considerably due to the identification of more causative genes and increased awareness from medical/scientific publications and media stories. Fewer than 30 cases are known in the UK [Authors, personal observation], giving an estimated prevalence of one in a million.
3. Evaluation Strategy to Identify the Genetic Cause of Congenital Insensitivity to Pain
The diagnosis of a specific Mendelian form of congenital insensitivity to pain is established in a proband with a suggestive history and/or presenting findings and a heterozygous pathogenic (or likely pathogenic) variant in SCN11A or biallelic pathogenic (or likely pathogenic) variants in one of the other genes listed in Table 1. The diagnosis can be difficult to establish on clinical grounds alone before age five years.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) The identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of targeted gene testing (multigene panel, single-gene testing) and genomic testing (comprehensive genomic sequencing).
Targeted gene testing requires the clinician to develop a hypothesis as to which specific gene(s) are likely to be involved, whereas genomic testing does not. Targeted testing is feasible based on phenotype in anyone older than approximately age five years (because of the difficulties of assessing subtle problems of intellectual developmental, sweating, temperature sensing, and autonomic features in infants and young children), with the exception of SCN11A (see Serial single-gene testing following).
Serial single-gene testing. The phenotype may guide the choice of which gene(s) to analyze first. Consider performing sequence analysis of:
SCN11A first in a newborn with severe intestinal hypomotility.
SCN9A first in an individual with normal intelligence who has anosmia.
PRDM12 first in an individual with normal intelligence, staphylococcal infections, and hypohidrosis.
Because some individuals with NTRK1-CIP and NGF-CIP have minimal learning problems, sequence analysis of these two genes should be considered next if molecular genetic testing of SCN9A and PRDM12 yield no pathogenic variants.
NTRK1 and NGF first in an individual with evidence of learning problems or late development, staphylococcal infections, and hypohidrosis. Unexplained fever due to anhidrosis (the inability to sweat) is a characteristic and often initial feature (more so in hot climates).
If no pathogenic variant is found in SCN11A or ZFHX2 through sequence analysis OR if no or only one pathogenic variant is found through sequence analysis of the remainder of the genes listed in Table 1, gene-targeted deletion/duplication analysis should be considered.
Note: Whole-gene deletions have been reported in individuals with NGF-CIP [Fitzgibbon et al 2009] and SCN9A-CIP [Author, personal observation].
A multigene panel that includes genes for CIP and other genes of interest (see Table 2) may be considered. Note: The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Comprehensive genomic testing (when available) including exome and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
4. Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with congenital insensitivity to pain (CIPA), the evaluations summarized Table 3 (if not performed as part of the evaluation that led to the diagnosis) should be considered. Based on the underlying genetic cause and/or specific pathogenic variant, not all evaluations need to be done in every individual with CIPA but should be guided by the specific phenotypic features seen in association with the genes discussed in Table 1.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Congenital Insensitivity to Pain Disorders
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Skin 1
| Physical exam of the skin | Assess for dry skin & palmoplantar hyperkeratosis (often assoc w/cracking); determine if person is using a skin moisturizer daily. |
|
Regulation of body temperature
| Inquire about history of hyperthermia or hypothermia. | |
|
Insensitivity to pain
|
Multiple unintentional injuries
| Physical exam of the whole body | Assess for bruises, cuts, burns, biting & Staphylococcus aureus infections. |
|
Orthopedic injuries
| Exam of the bones & joints by an orthopedist | Assess for fractures; avascular necrosis; septic arthritis / osteomyelitis; self-mutilation; joint subluxation; Charcot neuroarthropathy; leg length discrepancy; scoliosis. Consider baseline radiography of lower spine, hips, knees, & ankles, if ambulatory.
|
|
Dental risks for injury
| Exam for oral lesions | Assess for traumatic lingual injuries, burns, self-biting, & auto-extraction of teeth, as well as overall dental health. |
|
Neuropathic keratitis
| Ophthalmologic exam | Assess for superficial punctate keratopathy, as well as corneal ulceration / perforation / infection. |
|
Developmental delay 2
| Neurologic exam & standardized tests for developmental milestones | Assess for development delay, as well as intellectual disability, incl defects in conceptual thinking & abstract reasoning. |
|
Behavior problems 3
| Formal eval of cognitive & adaptive functions | Assess for social behaviors & emotional disturbances; ADHD. |
|
Anosmia 4
| Inquire about sense of smell. | Affected persons may be unaware of body odor & rancid food. |
|
Genetic counseling
| By genetics professionals 5 | To obtain a pedigree & inform affected persons & families re nature, MOI, & implications of CIPA in order to facilitate medical & personal decision making |
Family support
& resources
| Assess need for:
| |
ADHD = attention-deficit/hyperactivity disorder; MOI = mode of inheritance
- 1.
Anhidrosis is more common in individuals with pathogenic variants in NTRK1, ZFHX2, and biallelic null variants in NGF; hyperhidrosis may be present in those with the c.2432T>C (p.Leu811Pro) pathogenic variant in SCN11A; pruritis is typically seen in those with pathogenic variants in SCN11A.
- 2.
Most often associated with pathogenic variants in CLTCL1, NGF, NTRK1, and SCN11A.
- 3.
Most often associated with pathogenic variants in NTRK1.
- 4.
Most often associated with pathogenic variants in SCN9A.
- 5.
Medical geneticist, certified genetic counselor, or certified advanced genetic nurse.
Treatment of Manifestations
No consensus treatment or surveillance guidelines have been developed.
Treatment is supportive and is best provided by specialists in pediatrics, orthopedics, dentistry, ophthalmology, and dermatology (see Congenital Insensitivity to Pain with Anhidrosis).
Table 4.
Treatment of Manifestations in Individuals with Congenital Insensitivity to Pain
View in own window
| Manifestation/Concern | Treatment | Considerations/Other |
|---|
|
Dental & oral lesions
| Tooth extraction &/or filing (smoothing) of sharp incisal edges [Bodner et al 2002]; use of a mouth guard [Hutton & McKaig 2010] | |
|
Bone fractures
| Standard treatment w/careful & regular review, assuming healing may not occur, & low threshold for repeat radiological imaging until normal | Treatment w/an external fixator may → potentially serious infectious complications. |
|
Bone & joint deformity
| Corrective osteotomy | Prolonged & intensive monitoring is necessary to avoid deformity or incomplete healing. |
|
Leg length discrepancy
| Shoe lift or epiphysiodesis [Bar-On et al 2002] | The value of surgical intervention needs to be weighed against nonsurgical approaches incl close monitoring [Kim et al 2013]. |
Corneal ulceration/
perforation/infection
| Standard treatment w/careful & regular review, assuming healing may not occur | Corneal transplants will have increased risk of failure. |
|
Dry eyes
| Lubricating eye drops or ointments | Surgical treatment of neurotrophic keratitis has not been successful [Yagev et al 1999]. |
|
Long-standing infections
| Wide surgical debridement | Prevention may be difficult but is important (See Prevention of Primary Manifestations and Prevention of Secondary Complications.) |
|
Ulcerating foot lesions
| Standard treatment w/careful & regular review, assuming healing may not occur | Appropriate footwear & ankle support & periods of non-weight-bearing may be appropriate. |
|
Hyperthermia
| Direct cooling in bath or w/cooling blanket | Control of environmental temperatures is essential. |
|
Hypothermia
| Warming by blanket |
|
Skin dryness & cracking
| Topical moisturizer (lotion or cream) | Untreated dry skin can → skin infections, which ↑ risk for serious infections (cellulitis or osteomyelitis). |
Developmental delay /
Intellectual disability
| See Developmental Delay / Intellectual Disability Management Issues. | |
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Developmental delay / intellectual disability educational issues may be seen in those with CTCL1-CIP, NGF-CIP, NTRK-CIP, or SCN11A-CIP.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed.
Ages 5-21 years
In the US, an IEP based on the individual's level of function should be developed by the local public school district. Affected children are permitted to remain in the public school district until age 21.
Discussion about transition plans including financial, vocation/employment, and medical arrangements should begin at age 12 years. Developmental pediatricians can provide assistance with transition to adulthood.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Social/Behavioral Concerns
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications when necessary.
Irritability, hyperactivity, impulsivity, and acting-out behaviors typically improve with age.
Prevention of Primary Manifestations
Table 5.
Prevention of Primary Manifestations in Individuals with Congenital Insensitivity to Pain
View in own window
| Manifestation/Concern | Prevention | Considerations/Other |
|---|
|
Injuries occurring around the home
| Stair gates; soft-round edging on tables & protruding objects; guard all heating devices; close supervision of younger children in the kitchen | |
|
Injuries occurring at school
| Inform personnel at school of diagnosis; seek help when accident occurs but child does not seem hurt. | |
|
Self-inflicted injuries
| Education of affected individuals about their condition. | Communicating w/other families of individuals w/CIP (especially affected adults) |
|
Corneal abrasion
| At least annual ophthalmologic eval; artificial tears | Artificial tears are particularly helpful to those w/PRDM12-CIP. All persons w/congenital corneal anesthesia have had SCN9A-CIP [Author, personal observation].
|
|
Staphylococcus aureus infections
| Good hand hygiene & care; use of antiseptic soaps; early use of topical antibiotics Investigation of swollen joints, limping, & limb underuse for infection by radiograph & C-reactive protein
| Affects those w/NTRK1-, NGF-, CLTCL1-, & PRDM12-CIP; infections are specific for S aureus only. |
Prevention of Secondary Complications
Table 6.
Prevention of Secondary Manifestations in Individuals with Congenital Insensitivity to Pain
View in own window
| Manifestation/Concern | Prevention | Considerations/Other |
|---|
|
Inability to use pain as indicator in diagnosing or assessing injury severity
| Laminated information letters/MediAlert bracelets | Consider providing laminated letter confirming diagnosis, stating pathogenic variant(s), & giving advice on diagnosis & treatment. |
|
Osteomyelitis of mandible
| Early treatment of dental caries & periodontal disease | Regular dental exams & restriction of sweets |
|
Bone & joint injury due to strenuous activity when individual has poor baseline conditioning
| Activities that lead to increased strength, balance, & body awareness | Dancing (particularly ballet), swimming, cycling, & non-traumatic martial arts may be considered. |
|
Inadequate sedation in postoperative period may trigger unexpected movement, causing secondary injury.
| Adequate sedation during procedures | Tachycardia & hypertension in postoperative period should raise consideration of the possibility of inadequate sedation. |
|
Hyper- or hypothermia
| Careful monitoring of temperature during perioperative period | |
Surveillance
In addition to regular evaluations by a pediatrician and dermatologist (to assess and advise on skin infections/injuries) the measures in Table 7 are recommended.
Table 7.
Recommended Surveillance for Individuals Congenital Insensitivity to Pain
View in own window
| Manifestation/Concern | Evaluation | Frequency/Comment |
|---|
|
Dental caries / Tooth damage
| Dental care | Regular exams (at least every 6 mos) |
|
Early injuries
| Eval by parents & caregivers for signs of unrecognized injury | Daily |
|
Bone health
| Prompt investigation & treatment of orthopedic consequences of CIP by named orthopedic surgeon | At least annually; more frequently depending on bony injuries |
|
Corneal damage
| Ophthalmology eval | At least annually; more frequently as indicated |
|
Hyper- or hypothermia
| Monitoring of body temperature may allow timely treatment of hyper- or hypothermia. | As needed |
|
Charcot joints
| Expert orthopedist assessment incl radiography of lower spine, hips, knees, & ankles | Every 1-3 yrs |
Agents/Circumstances to Avoid
Avoid the following:
Jumping, high-impact/contact sports, pastimes and jobs that involve the potential for blunt injury or severe bone and joint trauma
The paucity of males with CIP who are older than age 20 years correlates with behaviors fueled by inability to feel pain (e.g., greater risk taking, deliberately picking fights, participation in extreme sporting events).
Hot or cold environments; hot or cold foods, hot showers or baths; heating blankets, particularly in the perioperative period
Pregnancy Management
Women with CIP are able to become pregnant and bear children normally.
Obstetric staff must be made aware of the diagnosis of CIP. Labor progresses normally, but will be painless, while other senses (stretch and touch) are intact. A delay in detecting pelvic fractures in an affected woman in the postnatal period has been reported [Wheeler et al 2014].
Other
Individuals with CIP typically learn that others have pain and tend to respond to others' pain normally. They often learn to simulate having pain in appropriate situations, e.g., being tackled during football.
The possibility that naloxone may temporarily relieve CIP analgesia has been suggested [Minett et al 2015]. While this medication could be of use in detecting the source of injury/illness in an affected individual, it may also expose the affected person to widespread pain from accumulated injuries.
5. Genetic Risk Assessment
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Congenital insensitivity to pain (CIP) is inherited in an autosomal recessive manner, with the exceptions of SCN11A-CIP and ZFHX2-CIP, which are inherited in an autosomal dominant manner.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of an affected child are obligate heterozygotes (i.e., carriers of one CIP-related pathogenic variant).
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic and are not at risk for developing the disorder.
Offspring of a proband. The offspring of an individual with CIP are obligate heterozygotes (carriers) for a pathogenic variant in a CIP-related gene.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a CIP-related pathogenic variant.
Carrier detection. Carrier testing for at-risk relatives requires prior identification of the CIP-related pathogenic variants in the family.
Autosomal Dominant Inheritance (SCN11A-CIP and ZFHX2-CIP) – Risk to Family Members
See SCN11A and ZFHX2 in Table 1 for information on these very rare conditions.
Parents of a proband
Some individuals diagnosed with SCN11A-CIP have an affected parent.
Some individuals diagnosed with SCN11A-CIP have the disorder as the result of a de novo pathogenic variant. Information on the frequency of de novo pathogenic variants is currently very limited.
Inheritance in the one family reported to date with
ZFHX2-CIP was autosomal dominant, with six affected individuals in three generations [
Habib et al 2018].
Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant.
If the pathogenic variant found in the proband cannot be detected in leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent. Though theoretically possible, no instances of germline mosaicism have been reported.
Sibs of a proband
The risk to the sibs of the proband depends on the genetic status of the proband's parents.
If a parent of the proband is affected, the risk to the sibs is 50%. Based on currently available, but limited, information, SCN11A- and ZFHX2-CIP appear to be completely penetrant. Phenotypic variability within families has not been reported.
If the parents have been tested for the pathogenic variant identified in the proband and:
A parent of the proband has the pathogenic variant, the risk to the sibs of inheriting the variant is 50%. Based on currently available (limited) information, SCN11A- and ZFHX2-CIP appear to be completely penetrant without familial phenotypic variability.
If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
If the parents have not been tested for the pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. The sibs of a proband with clinically unaffected parents are still at increased risk for CIP because of the theoretic possibilities of reduced penetrance in a parent or parental germline mosaicism [
Rahbari et al 2016].
Offspring of a proband. Each child of an individual with SCN11A- or ZFHX2-CIP has a 50% chance of inheriting the pathogenic variant. Based on currently available (limited) information, SCN11A- and ZFHX2-CIP appear to be completely penetrant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the SCN11A or ZFHX2 pathogenic variant or is clinically affected, the parent's family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the CIP-related pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal and preimplantation genetic testing. While most health care professionals would consider use of prenatal and preimplantation genetic testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
National Library of Medicine Genetics Home Reference
Tomorrow: The Japan Association of Patients with Congenital Insensitivity to Pain with Anhidrosis (CIPA)
Provides information about CIPA (HSAN IV) in Japanese
Kitami 8-15-35-307
Tokyo 157-0067
Japan
Phone: 03-5761-2860
Fax: 03-5761-2861
Email: cipa@tomorrow.or.jp
Chapter Notes
Author Notes
AP+CGW run a clinical and advice service for diagnosis and management of CIP.
Acknowledgments
We thank families with congenital insensitivity to pain and colleagues for advice in writing this paper.
References
Literature Cited
Bar-On
E, Weigl
D, Parvari
R, Katz
K, Weitz
R, Steinberg
T. Congenital insensitivity to pain: orthopaedic manifestations.
J Bone Joint Surg Br.
2002;84:252-7
[
PubMed: 11922368]
Bodner L, Woldenberg Y, Pinsk V, Levy J. Orofacial manifestations of congenital insensitivity to pain with anhidrosis: a report of 24 cases. ASDC J Dent Child. 2002;69:293-6, 235 [
PubMed: 12613315]
Carvalho
OP, Thornton
GK, Hertecant
J, Houlden
H, Nicholas
AK, Cox
JJ, Rielly
M, Al-Gazali
L, Woods
CG. A novel NGF mutation clarifies the molecular mechanism and extends the phenotypic spectrum of the HSAN5 neuropathy.
J Med Genet.
2011;48:131-5.
[
PMC free article: PMC3030776] [
PubMed: 20978020]
Chen
Y-C, Auer-Grumbach
M, Matsukawa
S, Zitzelsberger
M, Themistocleous
AC, Strom
TM, Samara
C, Moore
AW, Cho
LT-Y, Young
GT, Weiss
C, Schabhüttl
M, Stucka
R, Schmid
AB, Parman
Y, Graul-Neumann
L, Heinritz
W, Passarge
E, Watson
RM, Hertz
JM, Moog
U, Baumgartner
M, Valente
EM, Pereira
D, Restrepo
CM, Katona
I, Dusl
M, Stendel
C, Wieland
T, Stafford
F, Reimann
F, von Au
K, Finke
C, Willems
PJ, Nahorski
MS, Shaikh
SS, Carvalho
OP, Nicholas
AK, Karbani
G, McAleer
MA, Cilio
MR, McHugh
JC, Murphy
SM, Irvine
AD, Jensen
UB, Windhager
R, Weis
J, Bergmann
C, Rautenstrauss
B, Baets
J, De Jonghe
P, Reilly
MM, Kropatsch
R, Kurth
I, Chrast
R, Michiue
T, Bennett
DLH, Woods
CG, Senderek
J. Transcriptional regulator PRDM12 is essential for human pain perception.
Nat Genet.
2015;47:803-8.
[
PMC free article: PMC7212047] [
PubMed: 26005867]
Cox
JJ, Reimann
F, Nicholas
AK, Thornton
G, Roberts
E, Springell
K, Karbani
G, Jafri
H, Mannan
J, Raashid
Y, Al-Gazali
L, Hamamy
H, Valente
EM, Gorman
S, Williams
R, McHale
DP, Wood
JN, Gribble
FM, Woods
CG. An SCN9A channelopathy causes congenital inability to experience pain.
Nature.
2006;444:894-8.
[
PMC free article: PMC7212082] [
PubMed: 17167479]
Cox
JJ, Sheynin
J, Shorer
Z, Reimann
F, Nicholas
AK, Zubovic
L, Baralle
M, Wraige
E, Manor
E, Levy
J, Woods
CG, Parvari
R. Congenital insensitivity to pain: novel SCN9A missense and in-frame deletion mutations.
Hum Mutat.
2010;31:E1670-86.
[
PMC free article: PMC2966863] [
PubMed: 20635406]
Einarsdottir
E, Carlsson
A, Minde
J, Toolanen
G, Svensson
O, Solders
G, Holmgren
G, Holmberg
D, Holmberg
M.
A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception.
Hum Mol Genet.
2004;13:799-805.
[
PubMed: 14976160]
Fitzgibbon
GJ, Kingston
H, Needham
M, Gaunt
L. Haploinsufficiency of the nerve growth factor beta gene in a 1p13 deleted female child with an insensitivity to pain.
Dev Med Child Neurol.
2009;51:833-7.
[
PubMed: 19183217]
Goldberg
YP, MacFarlane
J, MacDonald
ML, Thompson
J, Dube
MP, Mattice
M, Fraser
R, Young
C, Hossain
S, Pape
T, Payne
B, Radomski
C, Donaldson
G, Ives
E, Cox
J, Younghusband
HB, Green
R, Duff
A, Boltshauser
E, Grinspan
GA, Dimon
JH, Sibley
BG, Andria
G, Toscano
E, Kerdraon
J, Bowsher
D, Pimstone
SN, Samuels
ME, Sherrington
R, Hayden
MR. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations.
Clin Genet.
2007;71:311-19.
[
PubMed: 17470132]
Habib
AM, Matsuyama
A, Okorokov
AL, Santana-Varela
S, Bras
JT, Aloisi
AM, Emery
EC, Bogdanov
YD, Follenfant
M, Gossage
SJ, Gras
M, Humphrey
J, et al.
A novel human pain insensitivity disorder caused by a point mutation in ZFHX2.
Brain.
2018;141:365-76.
[
PMC free article: PMC5837393] [
PubMed: 29253101]
Huang
J, Vanoye
CG, Cutts
A, Goldberg
YP, Dib-Hajj
SD, Cohen
CJ, Waxman
SG, George
AL
Jr. Sodium channel Na
v1.9 mutations associated with insensitivity to pain dampen neuronal excitability.
J Clin Invest.
2017;127:2805-14
[
PMC free article: PMC5490760] [
PubMed: 28530638]
Hutton
A, McKaig
S. The dental management of a child with congenital insensitivity to pain.
Dent Update.
2010;37:180-2.
[
PubMed: 20491220]
Iftikhar
S, Javed
MA. Seeing is not always believing: congenital insensitivity to pain with anhidrosis mimicking leprosy.
Mayo Clin Proc.
2013;88:e153-4
[
PubMed: 24290131]
Indo
Y.
Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): mutations and polymorphisms in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor.
Hum Mutat.
2001;18:462-71.
[
PubMed: 11748840]
Indo
Y, Tsuruta
M, Hayashida
Y, Karim
MA, Ohta
K, Kawano
T, Mitsubuchi
H, Tonoki
H, Awaya
Y, Matsuda
I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis.
Nat Genet.
1996;13:485-8.
[
PubMed: 8696348]
Leipold
E, Liebmann
L, Korenke
GC, Heinrich
T, Giesselmann
S, Baets
J, Ebbinghaus
M, Goral
RO, Stödberg
T, Hennings
JC, Bergmann
M, Altmüller
J, Thiele
H, Wetzel
A, Nürnberg
P, Timmerman
V, De Jonghe
P, Blum
R, Schaible
HG, Weis
J, Heinemann
SH, Hübner
CA, Kurth
I. A de novo gain-of-function mutation in SCN11A causes loss of pain perception.
Nat Genet.
2013;45:1399-1404.
[
PubMed: 24036948]
Levy Erez
D, Levy
J, Friger
M, Aharoni-Mayer
Y, Cohen-Iluz
M, Goldstein
E.
Assessment of cognitive and adaptive behaviour among individuals with congenital insensitivity to pain and anhidrosis.
Dev Med Child Neurol.
2010;52:559-62.
[
PubMed: 20089052]
King
MK, Leipold
E, Goehringer
JM, Kurth
I, Challman
TD. Pain insensitivity: distal S6-segment mutations in Nav1.9 emerge as a critical hotspot.
Neurogenetics.
2017;18:179-81.
[
PubMed: 28289907]
Minett
MS, Pereira
V, Sikandar
S, Matsuyama
A, Lolignier
S, Kanellopoulos
AH, Mancini
F, Iannetti
GD, Bogdanov
YD, Santana Varela
S, Millet
Q, Baskozos
G, MacAllister
R, Cox
JJ, Zhao
J, Wood
JN. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7.
Nat Commun.
2015;6:8967.
[
PMC free article: PMC4686868] [
PubMed: 26634308]
Nahorski
MS, Al-Gazali
L, Hertecant
J, Owen
DJ, Borner
GH, Chen
YC, Benn
CL, Carvalho
OP, Shaikh
SS, Phelan
A, Robinson
MS, Royle
SJ, Woods
CG. A novel disorder reveals clathrin heavy chain-22 is essential for human pain and touch development.
Brain.
2015a;138:2147-60.
[
PMC free article: PMC4511860] [
PubMed: 26068709]
Nahorski
MS, Chen
YC, Woods
CG. New Mendelian disorders of painlessness.
Trends Neurosci.
2015b;38:712-24.
[
PubMed: 26549885]
Phatarakijnirund
V, Mumm
S, McAlister
WH, Novack
DV, Wenkert
D, Clements
KL, Whyte
MP. Congenital insensitivity to pain: fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1.9.
Bone.
2016;84:289-98.
[
PMC free article: PMC4755825] [
PubMed: 26746779]
Rahbari
R, Wuster
A, Lindsay
SJ, Hardwick
RJ, Alexandrov
LB, Turki
SA, Dominiczak
A, Morris
A, Porteous
D, Smith
B, Stratton
MR, Hurles
ME, et al.
Timing, rates and spectra of human germline mutation.
Nat Genet.
2016;48:126-33
[
PMC free article: PMC4731925] [
PubMed: 26656846]
Richards
S, Aziz
N, Bale
S, Bick
D, Das
S, Gastier-Foster
J, Grody
WW, Hegde
M, Lyon
E, Spector
E, Voelkerding
K, Rehm
HL, et al.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med.
2015;17:405-24.
[
PMC free article: PMC4544753] [
PubMed: 25741868]
Saini
AG, Padmanabh
H, Sahu
JK, Kurth
I, Voigt
M, Singhi
P. Hereditary sensory polyneuropathy, pain insensitivity and global developmental delay due to novel mutation in
PRDM12 gene.
Indian J Pediatr.
2017;84,332-3
[
PubMed: 28050684]
Shatzky
S, Moses
S, Levy
J, Pinsk
V, Hershkovitz
E, Herzog
L, Shorer
Z, Luder
A, Parvari
R. Congenital insensitivity to pain with anhidrosis (CIPA) in Israeli-Bedouins: genetic heterogeneity, novel mutations in the TRKA/NGF receptor gene, clinical findings, and results of nerve conduction studies.
Am J Med Genet.
2000;92:353-60.
[
PubMed: 10861667]
Spinsanti
G, Zannolli
R, Panti
C, Ceccarelli
I, Marsili
L, Bachiocco
V, Frati
F, Aloisi
AM. Quantitative real-time PCR detection of TRPV1-4 gene expression in human leukocytes from healthy and hyposensitive subjects.
Mol Pain.
2008;4:51.
[
PMC free article: PMC2588574] [
PubMed: 18983665]
Staudt
MD, Bailey
CS, Siddiqi
F. Charcot spinal arthropathy in patients with congenital insensitivity to pain: a report of two cases and review of the literature.
Neurosurg Rev.
2018;41:899-908.
[
PubMed: 28124176]
Weiss
J, Pyrski
M, Jacobi
E, Bufe
B, Willnecker
V, Schick
B, Zizzari
P, Gossage
SJ, Greer
CA, Leinders-Zufall
T, Woods
CG, Wood
JN, Zufall
F. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia.
Nature.
2011;472:186-90.
[
PMC free article: PMC3674497] [
PubMed: 21441906]
Yagev
R, Levy
J, Shorer
Z, Lifshitz
T. Congenital insensitivity to pain with anhidrosis: ocular and systemic manifestations.
Am J Ophthalmol.
1999;127:322-6.
[
PubMed: 10088743]
Zhang
S, Malik Sharif
S, Chen
YC, Valente
EM, Ahmed
M, Sheridan
E, Bennett
C, Woods
G. Clinical features for diagnosis and management of patients with PRDM12 congenital insensitivity to pain.
J Med Genet.
2016;53:533-5.
[
PMC free article: PMC4975812] [
PubMed: 26975306]