

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

Glycogen storage disease II, or Pompe disease, is a rare and often fatal autosomal recessive lysosomal storage disorder (LSD) caused by the dysfunction of the lysosomal enzyme acid alpha-glucosidase (GAA). Accumulation of GAA’s substrate, glycogen, causes enlargement of cellular lysosomes, adversely affecting many cells, especially heart and skeletal muscle tissues. The only FDA approved treatment for Pompe disease is enzyme replacement therapy, called Myozyme, which has significant limitations. Importantly, of the over 100 different mutations known to cause Pompe disease, many retain enzymatic activity in vitro, although the structural changes induced by mutants affect trafficking of the enzyme to the lysosome. Small molecule chaperones can be used to correct this trafficking defect. These compounds bind to the protein in the endoplasmic reticulum, accelerating the folding process and increasing their translocation to the lysosome, thereby reducing substrate accumulation. Several iminosugar inhibitors of GAA, such duvoglustat, are known to chaperone the translocation of mutant GAA proteins. However, their impact on substrate reduction may be limited by their continued inhibition of the target enzyme, as well as limited selectivity towards GAA. We have previously reported an inhibitor (ML201) and through further work, we have now also identified the first non-inhibitory small molecule chaperone of acid alpha glucosidase, ML247. Here, we demonstrate that ML247 enhances mutant enzyme translocation using Pompe patient-derived fibroblasts. ML247 displays reasonable pharmacokinetics and might serve as a pivotal first step in efforts to develop a non-inhibitory molecular chaperone for the treatment of Pompe disease.
Screening Center Name & PI: NIH Chemical Genomics Center, Christopher P. Austin
Chemistry Center Name & PI: NIH Chemical Genomics Center, Christopher P. Austin
Assay Submitter & Institution: Wei Zheng, NIH Chemical Genomics Center
PubChem Summary Bioassay Identifier (AID): 1473
Probe Structure & Characteristics
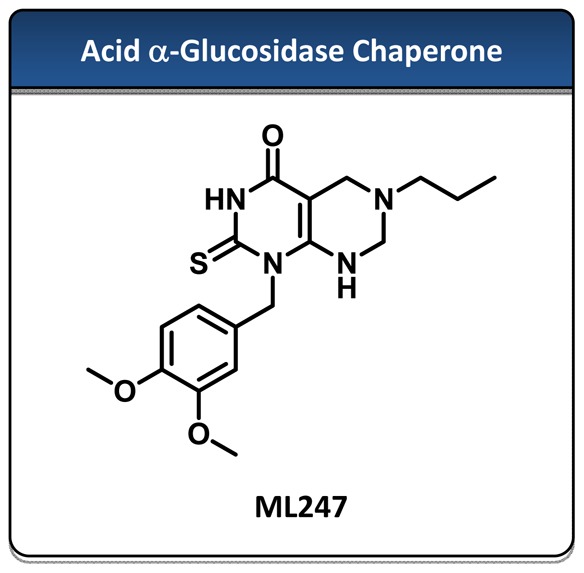
| CID/ML | Target Name | IC50/EC50 (nM) [SID, AID] | Anti-target Name(s) | IC50/EC50 (μM) [SID, AID] | Selectivity | Secondary Assay(s) Name: IC50/EC50 (μM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 44246403/ML247 | Acid α-Glucosidase | 2818 [SID 85267344, AID 2110] | β-Glucosidase α-Galactosidase | >57 μM [AID 540341] >57 μM [AID 540361] | >10-fold | 15 [SID 85267344, AID 602122] |
Recommendations for Scientific Use of the Probe
Mutations in the lysosomal enzyme acid alpha-glucosidase (GAA) are the underlying cause of Pompe disease. Here, we present a small molecule chaperone that can correct the misfolding and mistrafficking of disease-causing mutant protein. All previously described small molecule chaperones of GAA are inhibitors of the enzyme. Here, we disclose a new class of non-inhibitory, selective small-molecule GAA chaperones that increase the translocation of several GAA mutants in primary patient fibroblasts without inhibiting the translocated protein’s specific activity. These molecules increase the rate of hydrolysis in enzymatic assays, measured through several of the classical fluorescent substrates of GAA. The probe molecule displays reasonable PK properties and exposure in vivo and is an ideal candidate for further characterization of its ability to reduce substrate accumulation in vivo. However, it should be noted that ML247 is not the most potent molecule with chaperone activity. With a potency of 160 nM, CID 44246394 may be more appropriate for in vitro studies, especially mechanism of action studies. Unfortunately, it is expected to be rapidly metabolized in vivo.
1. Introduction
Pompe disease, also called glycogen storage disease type II or acid maltase deficiency is an autosomal recessive disorder caused by the deficiency or dysfunction of the lysosomal enzyme acid alpha-glucosidase (GAA).1 Epidemiological studies have estimated its frequency to be 1 in every 40,000 births.1,2 Functionally, GAA hydrolyzes terminal α-1,4 glucosidic linkages of glycogen in the lysosome. Mutations in this enzyme result in lysosomal enlargement due to glycogen accumulation, which is especially severe in cardiac and skeletal muscle, affecting breathing and mobility.1 The only therapy currently approved by the FDA for Pompe Disease is enzyme replacement therapy (Myozyme), which is recombinant GAA produced in a Chinese hamster ovary cell line3. Although Myozyme has been proven to be clinically efficacious in children, the development of infusion-related reactions is common, and the majority of the patients (89%) test positive for IgG antibodies to acid alpha-glucosidase, reducing its clinical utility.4 In addition, some health plans have refused to subsidize Myozyme for adult patients, because it is only approved for children and has a high annual cost of treatment ($300,000/year). Common observed adverse effects to Myozyme treatment are pneumonia, respiratory complications, infections, and fever. More serious reactions include heart and lung failure and allergic shock.5 These findings reinforce the need to develop new treatments for Pompe disease.
There are more than 100 different GAA mutations that can produce Pompe disease symptoms.6 Many of these mutant proteins retain their enzymatic activity in vitro, but they are not effectively transported to the lysosome. They start accumulating in the endoplasmic reticulum (ER), presumably due to their inability to fold properly or acquire the necessary shape to be targeted for transportation to the lysosome.7 Eventually, these mutants are tagged through ubiquitination and directed to the proteosome for degradation.
A general strategy for the treatment of lysosomal storage diseases (LSDs) has been to search for small molecule chaperones that are able to bind to the misfolded mutant enzymes and assist in their folding and transport to the lysosome. This improves trafficking of the mutant enzymes between the ER and the lysosome and reduces substrate accumulation and lysosome size.8 Paradoxically, almost all of the small molecule chaperones reported in the literature are also enzyme inhibitors, with the majority being iminosugars. One of these, duvoglustat (1-deoxynojirimycin, AT2220; Figure 1), is currently being tested in a phase II clinical trial as a pharmacological chaperone for Pompe disease by Amicus Therapeutics Inc.9

Figure 1
Previously described inhibitory GAA chaperones.
Iminosugars bind at the active site of the enzyme, mimicking the transition state of the glycolytic reaction. Binding of iminosugars to the active site stabilizes the protein in an active conformation and facilitates its transport to the lysosome. Several authors have reported the use of different iminosugar inhibitors as chaperone molecules for the correction of a number of lysosomal storage diseases (LDSs).8 Although the ability of iminosugars to act as protein chaperones has been well established, this strategy has several pitfalls. First, because of their mode of action as substrate mimetics, iminosugars tend to be poorly selective8, inhibiting several glycosidases simultaneously and producing many side effects. Second, patient responses have also been highly variable, due to population differences in the expression of membrane transporters, which affect how the iminosugars are actively transported through the gastrointestinal track and the blood-brain-barrier. Lastly and most importantly, due to their intrinsic inhibitory activity, the therapeutic window of these molecules can be small because of the narrow difference in concentration at which they are translocation inducers and inhibitors.8,10
Upon translocation to the lysosome, these orthosteric small molecule chaperones must be allowed to be displaced by the native substrate. This displacement depends on the rate of binding between substrate and inhibitor; therefore, weak inhibitors are preferred. In addition, their concentrations at the site of action also impact the equilibrium. For this reason, the dosing and schedule of these small molecule chaperones must be carefully tailored to achieve optimal exposures in vivo that favor enzyme translocation without reducing enzyme activity. All of these considerations make the development of iminosugar chaperones difficult; therefore, it is of interest to search for novel series of compounds with chaperone activity. In the past, our group has disclosed an alternative inhibitory series with GAA chaperone activity (Figure 1, ML201) with improved selectivity compared to the iminosugar inhibitors. Here, we describe the first non-inhibitory, selective small molecule chaperone of GAA, ML247.
2. Materials and Methods
The reagents and solvents were used as commercial anhydrous grade without further purification. The column chromatography was carried out over silica gel (100–200 mesh). 1H NMR spectra were recorded with a Bruker 400 MHz spectrometer from solutions in CDCl3 and DMSO-d6. Chemical shifts in 1H NMR spectra are reported in parts per million (ppm, δ) downfield from the internal standard Me4Si (TMS, δ = 0 ppm). Molecular weight confirmation was performed using an Agilent Time-Of-Flight Mass Spectrometer (TOF, Agilent Technologies, Santa Clara, CA). A 3 minute gradient from 4 to 100% Acetonitrile (0.1% formic acid) in water (0.1% formic acid) was used with a 4 minute run time at a flow rate of 1 mL/min. A Zorbax SB-C18 column (3.5 micron, 2.1 × 30 mm) was used at a temperature of 50 °C. Confirmation of molecular formula was confirmed using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
2.1. Assays
Detailed descriptions and protocols of all these assays listed in Table 1 can be found in the appendix. For further information, including data, please see http://pubchem.ncbi.nlm.nih.gov/, especially the summary assay for the project, AID 1473. Curves can be viewed by looking at the data and selecting the graph function in “outcome”.
Table 1
List of all assays screened.
2.2. Probe Chemical Characterization
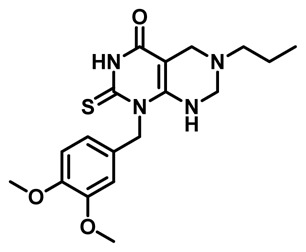
Probe Characterization of ML247
*Purity >95% as determined by LC/MS and 1H NMR analyses.
1-(3,4-dimethoxybenzyl)-6-propyl-2-thioxo-2,3,5,6,7,8-hexahydropyrimido[4,5-d]pyrimidin-4(1H)-one. 1H NMR (400MHz, DMSO-d6) δ 12.08 (s, 1H), 7.20 (s, 1H), 6.88 (d, J = 4.4 Hz, 2H), 6.65 (d, J = 8.0 Hz, 1H), 5.51 (br s, 2H), 3.92 (s, 2H), 3.70 (s, 6H), 3.39 (s, 2H), 2.32 (t, J = 7.2 Hz, 2H), 1.43 (q, J = 7.2 Hz, 2H), 0.84 (t, J = 7.2 Hz, 3H); HRMS (ESI) m/z calculated for [C18H24N4O3S + H]+ 376.1569, found 376.1573.
| Internal ID | MLS ID | SID | CID | ML | Type | Source |
|---|---|---|---|---|---|---|
| NCGC00183885-01 | MLS002699808 | 85267344 | 44246403 | ML247 | Probe | NCGC |
| NCGC00182856-01 | MLS000729403 | 85267296 | 1512045 | Analog | NCGC | |
| NCGC00183621-01 | MLS002699809 | 85267337 | 44246396 | Analog | NCGC | |
| NCGC00183442-01 | MLS002699810 | 85267314 | 44246374 | Analog | NCGC | |
| NCGC00183589-01 | MLS002699811 | 85267334 | 44246394 | Analog | NCGC | |
| NCGC00182855-01 | MLS002699812 | 85267104 | 1259573 | Analog | NCGC |

Figure 2List and structure of probe and analogs that have been submitted to the MLSMR
2.3. Probe Preparation
Scheme 1 shows the synthetic route used to ML247. Ammonium thiocyanate (27.58 mmol) was added to a stirred solution of the 3,4-dimthoxybenzyl amine, shown on the left (27.58 mmol) in bromobenzene (10 mL) at room temperature. The reaction mixture was heated at reflux temperature for 6 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The crude residue was purified by column chromatography (2–10% MeOH in CH2Cl2) to produce the corresponding thiourea above. The products were characterized by MS analysis and subjected to the next step without further purification; yield = 33%, MS (ESI) m/z [M + H]+ of 241 observed.

Scheme 1
Synthetic route to ML247.
The thiourea (2.08 mmol) and ethyl cyanoacetate (2.28 mmol) were then added to a solution of sodium (2.08 mmol) in ethanol (5 mL). The reaction mixture was heated at reflux for 4 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The crude residue was purified by column chromatography (1–3% MeOH in CH2Cl2) to afford the pyrimidinone shown above. The products were characterized by 1H NMR and MS analysis.
Yield = 31%; 1H NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 7.06 (s, 2H), 6.97 (s, 1H), 6.88–6.82 (m, 2H), 4.85 (s, 1H), 4.48 (br s, 2H), 3.73 (s, 3H), 3.70 (s, 3H), 2.84 (t, J = 8 Hz, 2H); HRMS (ESI) m/z calculated for [C14H17N3O3S + H]+ 307.0991, found 307.0993.
n-Propylamine (4.55 mmol) and formaldehyde (6.50 mmol) were added to a solution of the product amine from the previous step (3.25 mmol) in ethanol (20 mL). The reaction mixture was heated at reflux for 3 h, cooled to room temperature, and filtered through a sintered glass funnel to afford the final product, with a yield of 24%. The compound was characterized by 1H NMR and MS analysis, and HRMS.
3. Results
3.1. Summary of Screening Results
Initially, an enzyme assay was developed using recombinant GAA and a pro-fluorescent substrate, 4-methylumbelliferyl-α-D-glucopyranoside (4MU-α-Gluc). Upon hydrolysis by GAA, this substrate forms two products, glucose and 4-methyllumbelliferone (4MU). 4MU emits fluorescent signal at 440 nm when excited at 365 nm. This is a simple and homogenous assay that is suitable for HTS and can theoretically detect both activators and inhibitors. Using this assay, 199,176 compounds were tested (AID 2242). Signal to background ratios averaged 20-fold and plate Z’s averaged 0.7, indicating a robust assay. A counterscreen was run using recombinant alpha-galactosidase (GLA) and a similar pro-fluorescent 4MU-based substrate (AID 1467). Fluorescent compounds will appear to be enzyme activators in both assays, and by looking for selective activators of GAA, we hoped to identify non-inhibitory chaperones. We were not able to confirm any of the compounds from this initial screen as GAA activators.
Due to concerns that the recombinant, purified assay might be missing important protein factors, and the previous difficulty in finding compounds active in cell-based assays using hits against other purified lysosomal targets, the primary screen was reformatted to measure alpha-glucosidase specific activity from spleen homogenate. Signal to background ratios averaged 40-fold, and the average plate Z′ from the screen was 0.8 (Figure 4). By then, the MLSMR collection had grown, and 232,184 compounds were screened (AID 2112). The hit, CID 1512045, was cherry-picked from the MLSMR for confirmation based on selectivity against GLA in a similar screening format (AID 2107); this compound was not screened in the original primary screen and was the only compound whose activity was confirmed. The compound was then ordered from a commercial vendor and later re-synthesized in-house. This compound activates GAA in both the purified acid alpha-glucosidase assay and the tissue homogenate assay, with similar activity in both (Figure 5). CID 1512045 was not auto-fluorescent by spectral profiling and did not show activity against the related enzymes alpha-galactosidase and beta-glucosidase (glucocerebrosidase). Structure-activity relationship studies were then undertaken, leading to the identification of the probe compound, ML247.

Figure 4
Performance of primary qHTS screen. Both Z′ and %CV are very stable and consistent, indicating a robust screen. The pie chart depicts distribution of activity.

3.2. Dose-Response Curves for Probe
%20and%20the%20inhibitor%20CID%20176077%20(duvoglustat%2C%20green)%20in%20the%20alpha-glucosidase%20assay%20using%20purified%20enzyme%20against%20the%20resorufin%20tagged%20substrate.&p=BOOKS&id=153221_ml247f6.jpg "Click on image to zoom")
Figure 6Dose response curve for probe ML247 (red) and the inhibitor CID 176077 (duvoglustat, green) in the alpha-glucosidase assay using purified enzyme against the resorufin tagged substrate
3.3. Scaffold/Moiety Chemical Liabilities
It is not expected that this scaffold has any functional groups with serious chemical liabilities. Based on the results of microsomal stability studies (section 3.6), the probe has excellent stability in biological media. However, it is noteworthy that in the 48 hour compound stability study in PBS buffer, stability was below 75% (Figure 3). We were unable to determine whether the compound was slowly precipitating from solution or if there was hydrolysis of the thiourea moiety under the conditions tested. If use requires extended incubation times in aqueous media, compound stability should be closely monitored.

Figure 3
Stability of ML247 in DPBS (pH 7.4) at room temperature over 48 hours.
3.4. SAR Tables
For many compounds less active than the original hit, we were unable able to observe an inflection in the concentration-response at higher compound concentrations, and so we were unable to estimate an AC50 for those compounds. Furthermore, it was not possible to evaluate activity at higher than 100 μM due to solubility limitations of the compounds in DMSO. Instead, SAR was evaluated on the basis of a combination of AC50s and the maximum efficacy reached with each compound at 77 μM. That is the data reported below. Full concentration-response curves for every compound can be found in PubChem.
Tables 2–4 show the SAR of select compounds; the full set of analogs can be found in PubChem. SAR changes show that all core modifications that were synthesized (Table 2) failed to further activate hydrolysis of our fluorogenic substrate, and even replacement of the thiocarbonyl functional group by a carbonyl group diminished activity. Replacement of the pendant aromatic ring at position 1 with various alkyl chains also abolished activity. Several heteroaromatic rings pendant to position 1 were poorly active or greatly reduced the activity of the lead compound. Shortening the linker from two carbons to one, 16, is well tolerated and produces a small increment in efficacy. Optimization of the substitution pattern for the phenethyl group at position 1 (18) showed that electron donating di-substituted compounds tend to provide better efficacy. In addition, it can be seen that the most active molecules have very reasonable calculated log P and tPSA, and in general, increments of log P and reduction of tPSA correlated with improvements in activity. None of the compounds tested activate the hydrolysis of substrates for other glycosidase enzymes, such us alpha-Galactosidase A or Glucocerebrosidase (data not shown). Overall, the data shows a narrow SAR in which most modifications to the core of the molecule yielded diminished activation of hydrolysis. Full concentration-response curves for the most potent compounds 1, 10, 16, 18, 19 are shown in Figure 7. The best compound, 18, had an AC50 of 0.16 μM, and the probe molecule, ML247 (16), had a potency of 2.81 μM in this assay. We characterized in vitro activity against GAA, and then we proceeded to characterize the capacity of the most active compounds to translocate GAA to the lysosome in patient-derived fibroblasts as prima facie evidence of their capacity to chaperone GAA.
Table 3
SAR table with further modifications to the core’s alkyl substiuent. Activity is reported as in Table 2.
Table 4
SAR table with modifications to R1 of the molecule. Activity is reported as in Table 2.
Figure 7
Full concentration-response profiles of the best activators in the resorufin substrate and purified enzyme assay.
3.5. Cellular Activity
Chaperone Activity in Pompe Patient-derived Fibroblasts
To measure the capacity of our compounds to activate ER-lysosome translocation of GAA, we treated Pompe patient-derived fibroblasts with compound for five days, followed by immunostaining (GAA and Cathepsin D as lysosomal marker), wash, and image analysis. 13–14
Pompe fibroblasts are difficult to obtain, and in general, they grow slowly; they can only be expanded by a few passages. Two cell cultures derived from a single patient with the mutations p.Y455C/p.G638W were obtained. The residual specific activity of these patient-derived fibroblasts is around 3% of the activity displayed by fibroblast with wildtype GAA (data not shown).
Although the specific activity of GAA in fibroblasts (skin biopsies) is definitive for a Pompe disease diagnosis, fibroblasts do not express GAA in abundant amounts, and that expression decreases with increasing passage number, even for wildtype cells. Nevertheless, the present patient-derived cells had sufficient protein expression to enable us to characterize the chaperone activity of this series, which is done by examining the co-localization of GAA staining with lysosome marker staining (CathD). No lysosomal GAA staining can be seen in DMOS vehicle-treated Pompe cells. As a reference, in our hands, the GAA signal in wildtype fibroblast by passage 8 could be observed by staining in 10% of the cells.
The inhibitory chaperone duvoglustat is a non-selective iminosugar currently in clinical trials for Pompe disease. The chaperone activity of duvoglustat varies by mutation, and there is no previous literature indicating whether duvoglustat is active against p.Y455C/p.G638W GAA mutations. No chaperone activity was observed at compound concentrations of 1, 5, and 15 μM (data not shown). At 20 μM, translocation of GAA to the lysosome occurred in 3% of the cells. These values are in the same range as those reported for other GAA mutants, where duvoglustat increased GAA translocation at 20 μM. Duvoglustat does not increase the translocation of wildtype GAA (data not shown).
The initial hit compound 1 increases translocation of GAA to lysosomes in patient-derived fibroblasts. However, compound 16 gave the most robust responses, with around 10% translocation of mutant GAA to the lysosomes at 20 μM. Translocation could still be observed at 15 and 10 μM, but not at 5 μM. Comparison between the results obtained with duvoglustat and with compound 16, and the probe molecule ML247, show that at the same concentration, the probe molecule is superior (Figure 8).

3.6. Profiling Assays
Ultimately, compounds from this series need to be evaluated in vivo to demonstrate their therapeutic potential. The probe molecule from the series was selected for its potential to demonstrate activity in that setting; this depended not only on potency in vitro, but also activity in the cell-based translocation assay, microsomal stability, and to a lesser extent, Caco-2 permeability (Table 5). The probe compound, ML247, has excellent Caco-2 permeability (> 5cm−8/s) and robust stability, with >60% remaining after a 60 minute incubation with mouse liver microsomes. The addition of NADPH also had little effect on compound stability, indicating only very modest P450 mediated metabolism. In contrast, compounds 1, 18 and others (not shown) suffer from significant P450-mediated metabolism. Based on these results, the probe molecule, compound 16, was further studied in a single-dose pharmacokinetics experiment. At 50 mg/kg IP dosing, no acute toxicity was observed (Figure 10). Compound half-life was 2.75 hours in plasma, with concentrations in plasma, liver and heart all exceeding 10 μM at one hour. Reasonable exposure was generated by the compound to warrant exploratory testing in an in vivo efficacy model.
Table 5
In vitro profile of ML247.

Figure 10
In vivo exposure by ML247 following a single 50 mg/kg IP dose in mice. Further information can be found in the appendix.
4. Discussion
The ability of a small molecule to act as a protein chaperone that enhances protein translocation is normally measured using cell imaging assays. These assays are based on the immunostaining of a lysosomal hydrolase with specific antibodies and a lysosomal marker, such as LAMP2. These cell imaging assays are complicated and very labor intensive. They typically require at least five days of exposure to the chaperone compound, fixation of cells, treatment with antibodies, multiple wash steps, and finally high-content fluorescent imaging detection; they are not amenable to high throughput screening (HTS) and are unsuitable for supporting structure-activity relationship (SAR) studies. Thus, as a surrogate assay that is amenable to HTS, researchers in the field rely on enzymatic hydrolysis assays as the primary screening method to identify potential chaperones. Theoretically, this approach can identify activators and inhibitors of lysosomal hydrolases from a large compound library. Then, the chaperone activities of compounds are validated using the immunostaining assay.
The aim of this project was to identify non-inhibitory small molecule chaperones of GAA for the study and potential treatment of Pompe disease. These types of molecules have never been disclosed in literature, and from a therapeutic point of view, would be more desirable than the iminosugar chaperones currently in clinical trials. Non-inhibitory chaperones are allosteric binders by definition, because it is impossible to increase the rate of hydrolysis by competing at the active site with substrate, and should be more selective against other glycosidases, also possessing a wider therapeutic window than current treatments. We hypothesized that the present lack of such compounds was directly related to the screening methodology employed. Until now, the assays that have been used to measure enzyme modulation employed detergents to non-physiologically activate the enzyme. In retrospect, it may be the case that the optimization of the assay for obtaining maximal signal in these conditions might over-activate the enzyme into a conformation different than its physiological state, preventing the finding of activators. It has also been previously demonstrated that many glycosidases require allosteric activation by co-factors to be functional.12 We suggest the use of tissue homogenate preparations as the preferred approach to measure the functional activity of lysosomal enzymes. These preparations contain all of the necessary co-factors for enzyme activation in the natural environment, and as we demonstrate in our screening strategy, allow for the discovery of activating compounds.
The ability of an inhibitor to modulate the enzyme’s activity does not always correlate with its ability to induce and accelerate the folding and translocation of mutant enzyme. Therefore, inhibitors and activators can exist without chaperone activity, and vice-versa. If a particular compound series has both the capacity to modulate the activity of the enzyme and the ability to enhance its translocation, then compounds with a greater potency in modulating the enzymatic activity also usually show better translocation properties. In these cases, lower AC50 values imply higher affinity toward the enzyme, which should correlate with a greater capacity to induce a folded conformation and better translocation ability. However, even within a family of modulators, it is possible to increase the binding affinity of the series for the enzyme without incrementing its inhibitory or activatory activity; therefore, the best chaperone molecules within a series do not necessarily have to be the most potent modulators. Importantly, within series having modulatory and chaperone properties, the modulation of the enzyme activity can be used as an indirect method for measuring increments in affinity toward the enzyme, and therefore for improving the chaperone capacity of the series. From a therapeutic point of view, an ideal chaperone series should increase the translocation of the protein without inhibiting its enzymatic function.
It is also important to point out that from a structural point of view, lysosomal hydrolyses adopt slightly different conformations when accommodating different substrates in their active sites. Thus, the conformation that GAA adopts during the hydrolysis of the natural polymeric substrate glycogen could be substantially different than the one adopted for the hydrolysis of 4-methylumbelliferyl α-D-glucopyranoside or resorufin α-D-glucopyranoside. Allosteric small molecule chaperones able to bind with the enzyme and promote a particular conformation might influence the hydrolytic reaction of different substrates differently, depending on the substrates used. For this reason, activators of the hydrolysis of 4-methylumbelliferyl α-D-glucopyranoside do not necessarily have the same potency when they are evaluated using resorufin α-D-glucopyranoside or glycogen. The most relevant hydrolytic reaction can then be used for SAR studies to identify compounds with increased affinity toward the enzyme for testing in the chaperone assay.
After analyzing the expression of GAA from several different tissues, we decided to use human spleen homogenate as the source of the enzyme preparation, because of its high GAA specific activity. In contrast to other LSDs, there is not one dominant genotype in Pompe disease; therefore, we completed the primary screening using wildtype GAA. Only one series of activating molecules was confirmed. Additional LC/MS studies (Figure 11) showed that the activation of 4MU-α-glu hydrolysis was real and not a fluorescent artifact. Later studies with our hit (1), ML247, and other analogs showed that this series was selective toward modulating the activity of GAA. The capacity of this series to activate the hydrolysis of 4MU-α-glu did not reproduce upon the use of natural substrate glycogen (data not shown), but that measurement was carried out using an isolated enzyme preparation, because the reaction cannot be evaluated in tissue homogenate due to the high background of the product of the reaction (glucose) in that setting. Importantly, our molecules never displayed inhibitory activity in any of the enzymatic assays and conditions evaluated. In addition, we also showed that our probe molecule was able to increase the translocation of wildtype and mutant GAA to the lysosome. For the specific mutation used for this evaluation using Pompe fibroblasts, our probe molecule, ML247, displayed better translocation capacity (10% of the cells vs 3% for duvoglustat) than the known inhibitory chaperone duvoglustat, currently in phase II clinical trials.

Figure 11
LC/MS blue dye hydrolysis evaluation. Liquid chromatography traces of reaction mixtures shown. Peak identity assigned by mass spectrometry. Peak 1: 4MU-α-glu, peak 2: 4MU, peak 3: hit compound 1.
Finally, SAR changes show that all core modifications that were synthesized (Table 2) failed to further activate hydrolysis of our fluorogenic substrate, and even replacement of the thiocarbonyl functional group by a carbonyl group diminished activity. The SAR data also showed a low tolerance for most modifications, but we did manage to improve the AC50 16-fold of our lead compound (section 3.4 SAR). In addition, it can be seen that the most active molecules have very reasonable calculated log P and tPSA, and in general, increments of log P and reduction of tPSA correlated with improvements in activity. Moreover, microsomal stability and Caco-2 permeability studies prompted us to carry out in vivo pharmacokinetic evaluation (3.6 Profiling Assays). We observed that our probe molecule displayed reasonable levels and exposure in relevant tissues for the disease (heart, liver), and warrants further study of its efficacy.
In conclusion, we present for the first time a non-inhibitory chaperone series with capacity to translocate GAA mutants with improved activity in regards to the current clinical trial compound duvoglustat. Pharmacokinetic properties and therapeutic window considerations place our probe molecule as an excellent candidate for further advancement studies toward the treatment of Pompe disease.
4.1. Comparison to Existing Art and How the New Probe is an Improvement
Table 6Comparison of ML247 to prior art



All of the small molecule chaperones reported in the literature are enzyme inhibitors, with the majority being iminosugars. One of these, duvoglustat, is currently being tested in a phase II clinical trial as a pharmacological chaperone for Pompe disease by Amicus Therapeutics Inc.9 Iminosugars bind at the active site of the enzyme, mimicking the transition state of the glycolytic reaction. Binding of iminosugars to the active site stabilizes the protein in an active conformation and facilitates its transport to the lysosome. Although the ability of iminosugars to act as protein chaperones has been well established, this strategy has several pitfalls. First, because of their mode of action as substrate mimetics, iminosugars tend to be poorly selective8, inhibiting several glycosidases simultaneously and producing many side effects. Second, patient responses have also been highly variable, due to population differences in the expression of membrane transporters, which affect how the iminosugars are actively transported through the gastrointestinal track and the blood-brain-barrier. Lastly and most importantly, due to their intrinsic inhibitory activity, the therapeutic window of these molecules can be small because of the narrow difference in concentration at which they are translocation inducers and inhibitors.8,10
4.2. Mechanism of Action Studies
To be sure that these compounds increase the hydrolytic rate of 4MU-α-glu, and do not interfere with the assay readout, we decided to follow the reaction directly using LC/MS with the initial hit, 1. Figure 11 shows how the product of the reaction increased when we incremented the concentration of compound 1. No 4MU product was produced with compound in absence of enzyme (data not shown).
4.3. Planned Future Studies
As illustrated in our translocation studies, several of our best compounds, including our probe molecule ML247, are currently able to increase the trafficking of GAA between the ER and the lysosome (Figure 9). ML247 is not an inhibitor, displaying good pharmacokinetics with proper distribution and very reasonable exposure. We believe that this molecule is a good candidate for further studies in Pompe disease. Unfortunately, at present, there are no commercial point mutant mouse models of Pompe disease where chaperone molecules can be evaluated in vivo. Nevertheless, Amicus Pharmaceuticals has been able to advance an iminosugar inhibitor chaperone to phase II clinical trials using their proprietary approach. Another direction to pursue is testing the effect on compound treatment on wildtype animals, because the probe molecule is able to increase the translocation of wildtype GAA. We hope that the compound might generate a measurable increase in specific activity in disease-related tissues, allowing us to evaluate the therapeutic value of this molecule. We have also published our results15 and hope that our findings will be of interest to the community at large and will lead to further studies.

Figure 9
Two representative fields showing profound restoration of lysosomal GAA in patient-derived fibroblasts (blue arrows), upon five day incubation with 20 μM probe compound ML247. Coloring scheme is the same as Figure 8.
5. References
- 1.
- Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. McGraw–Hill; New York: 2001. pp. 3389–3420.
- 2.
- Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN, Codd WJ, Hanna B, Alcabes P, Raben N, Plotz P. Am J Med Genet. 1998;79:69–72. [PubMed: 9738873]
- 3.
- a. Kishnani PS, Corzo D, Nicolino M, et al. Neurology. 2007;68:99–109. [PubMed: 17151339]
b. http://www.myozyme.com/ - 4.
- 5.
- 6.
- a. Raben N, Plotz P, Byrne BJ. Curr. Mol Med. 2002;2:145–166. [PubMed: 11949932]
b. http://www.hgmd.cf.ac.uk. - 7.
- a. Montalvo AL, Cariati R, Deganuto M, Guerci V, Garcia R, Ciana G, Bembi B, Pittis MG. Mol. Genet. Metab. 2004;81:203–208. [PubMed: 14972326]
b. Hermans MM, van Leenen D, Kroos MA, Beesley CE, Van der Ploeg AT, Sakuraba H, Wevers R, Kleijer W, Michelakakis H, Kirk EP, Fletcher J, Bosshard N, Basel-Vanagaite L, Besley G, Reuser AJ. Hum. Mutat. 2004;23:47–56. [PubMed: 14695532]
c. Reuser AJ, Kroos M, Willemsen R, Swallow D, Tager JM, Galjaard H. J. Clin. Invest. 1987;79:1689–1699. [PMC free article: PMC424503] [PubMed: 3108320]
d. Reuser AJ, Kroos M, Oude Elferink RP, Tager JM. J. Biol. Chem. 1985;260:8336–8341. [PubMed: 3159730] - 8.
- a. Beck M. Human Genetics. 2007;121:1–22. [PubMed: 17089160]
b. Horne G, Wilson FX, Tinsley J, Williams DH, Storer R. Drug Discov Today. 2011;16(3–4):107–118. [PubMed: 20817006] - 9.
- a. Parenti G, Zuppaldi A, Pittis GM, Tuzzi MR, Annunziata I, Meroni G, Porto C, Donaudy F, Rossi B, Rossi M, Filocamo M, Donati A, Bembi B, Ballabio A, Andria G. Mol Ther. 2007;15:508–514. [PubMed: 17213836]
b. Okumiya T, Kroos MA, Vliet LV, Takeuchi H, Van der Ploeg AT, Reuser AJ. Mol. Genet Metab. 2007;90:49–57. [PubMed: 17095274] - 10.
- Jian-Qiang F, Satoshi I. The FEBS J. 2007;274:4962–71. [PubMed: 17894781]
- 11.
- Motabar O, Shi Z, Goldin E, Liu K, Southall N, Sidransky E, Austin CP, Griffiths GL, Zheng Z. Anal. Biochem. 2009;390:79–84. [PMC free article: PMC2737366] [PubMed: 19371716]
- 12.
- a. John M, Wendeler M, Heller M, Sandhoff K, Kessler H. Biochemistry. 2006;45:5206–5216. [PubMed: 16618109]
b. Zwerschke W, Mannhardt B, Massimi P, Nauenburg S, Pim D, Nickel W, Banks L, Reuseri AJ, Jansen-Dürr P. J. Biol. Chem. 2000;275:9541. [PubMed: 10734102] - 13.
- Parenti G, Zuppaldi A, Gabriela PM, Rosaria TM, Annunziata I, Meroni G, Porto C, Donaudy F, Rossi B, Rossi M, Filocamo M, Donati A, Bembi B, Ballabio A, Andria G. Mol Ther. 2007;15(3):508–14. [PubMed: 17213836]
- 14.
- Okumiya T, Kroos MA, Van Vliet L, Takeuchi H, Van der Ploeg AT, Reuser AJJ. Molecular Genetics Metabolism. 2007;90:49–57. [PubMed: 17095274]
- 15.
- Marugan JJ, Zheng W, Motabar O, Southall N, Goldin E, Sidransky E, Aungst RA, Liu K, Sadhukhan SK, Austin C. Eur J. Med Chem. 2010;45:1880–97. [PMC free article: PMC2892120] [PubMed: 20206419]
Appendix. Pharmacokinetics protocol and levels
| Study Design | |||||||
|---|---|---|---|---|---|---|---|
| Treatment Group | Treatment | No. of animals | Route of admin. | Dose Level (mg/kg) | Dose Conc. (mg/mL) | Dose Volume (mL/kg) | Time points |
| 1 | NCGC00183885-01 | 30 | IP | 50 | 5 | 10 | Predose, 0.083, 0.25, 0.5, 1, 2, 4, 8, 12 and 24 hr, plasma, brain, liver and heart collection |
| Test article | NCGC00183885-01 |
|---|---|
| Test system | C57BL/6 mice, 15–20 g, male, N=30, purchased from SLAC Laboratory Animal Co. LTD Qualification No.: SCXK(SH)2007-0005 20703 |
| Food status | Free access to food and water |
| Administration | IP: 50 mg/kg ( 10 mL/kg) in 10% DMAC+10% Tetraethylene glycol+ 10% Solutol HS 15+ 70% Water via lower left abdominal quadrant injection (N=30) |
| Blood collection | The animals were anesthetized with isoflurane and restrained manually at the designated time points. Approximately 300 μL of blood samples were taken from the animals into K2EDTA tube via cardiac puncture. Blood samples were put on ice and centrifuged to obtain plasma sample (2000 g, 5 min under 4 °C) within 15 minutes post sampling. |
| Heart collection | Perfusion: The animal was euthanized by exsanguination. The blunt syringe was hold in place with a hemostat in the left ventricle for systemic perfusion. Cut the right auricle open to allow the blood to flow out. Perfuse the animals with cold saline (around 20 mL per animal) for a few minutes until blood is flushed. After the perfusion, the tissues was collected: Heart collection: Grasp the heart using a pair of ring forceps and cut above the right and left atrium. Place the heart on a piece of gauze, and then cut from the base to the apex in four sections to expose all the ventricles and atrium, rinsed with cold saline, dried on filtrate paper, weighed and snap frozen by placing into dry-ice. |
| Liver collection | Holding the liver lobes with forceps, and a pair of dissection scissors was used to separate the liver. The whole liver was collected, rinsed with cold saline, dried on filtrate paper, weighed and snap frozen by placing into dry-ice. |
| Brain collection | A mid-line incision was made in the animals scalp and skin retracted. The skull overlying the brain was removed. The whole brain was collected, rinsed with cold saline, dried on filtrate paper, weighed and snap frozen by placing into dry-ice. |
| Sample storage and disposition | Plasma, brain, liver and heart samples were stored at approximately −80 °C until analysis. The backup samples will be discarded after one month unless requested. |
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- The pharmacological chaperone AT2220 increases the specific activity and lysosomal delivery of mutant acid alpha-glucosidase, and promotes glycogen reduction in a transgenic mouse model of Pompe disease.[PLoS One. 2014]The pharmacological chaperone AT2220 increases the specific activity and lysosomal delivery of mutant acid alpha-glucosidase, and promotes glycogen reduction in a transgenic mouse model of Pompe disease.Khanna R, Powe AC Jr, Lun Y, Soska R, Feng J, Dhulipala R, Frascella M, Garcia A, Pellegrino LJ, Xu S, et al. PLoS One. 2014; 9(7):e102092. Epub 2014 Jul 18.
- Discovery of a novel noniminosugar acid α glucosidase chaperone series.[J Med Chem. 2012]Discovery of a novel noniminosugar acid α glucosidase chaperone series.Xiao J, Westbroek W, Motabar O, Lea WA, Hu X, Velayati A, Zheng W, Southall N, Gustafson AM, Goldin E, et al. J Med Chem. 2012 Sep 13; 55(17):7546-59. Epub 2012 Aug 17.
- Duvoglustat HCl Increases Systemic and Tissue Exposure of Active Acid α-Glucosidase in Pompe Patients Co-administered with Alglucosidase α.[Mol Ther. 2017]Duvoglustat HCl Increases Systemic and Tissue Exposure of Active Acid α-Glucosidase in Pompe Patients Co-administered with Alglucosidase α.Kishnani P, Tarnopolsky M, Roberts M, Sivakumar K, Dasouki M, Dimachkie MM, Finanger E, Goker-Alpan O, Guter KA, Mozaffar T, et al. Mol Ther. 2017 May 3; 25(5):1199-1208. Epub 2017 Mar 22.
- Review 5-(4-(4-Acetylphenyl)piperazin-1-ylsulfonyl)indolin-2-one Analogs as Inhibitors of Acid alpha-Glucosidase for Potential Chaperone Treatment of Pompe Disease or Intervention for Diabetes Mellitus Type 2.[Probe Reports from the NIH Mol...]Review 5-(4-(4-Acetylphenyl)piperazin-1-ylsulfonyl)indolin-2-one Analogs as Inhibitors of Acid alpha-Glucosidase for Potential Chaperone Treatment of Pompe Disease or Intervention for Diabetes Mellitus Type 2.Xiao J, Marugan JJ, Zheng W, Motabar O, Southall N, Goldin E, Sidransky E, Liu K, Ferrer M, Austin CP. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review Phenotype variations in early onset Pompe disease: diagnosis and treatment results with Myozyme.[Adv Exp Med Biol. 2009]Review Phenotype variations in early onset Pompe disease: diagnosis and treatment results with Myozyme.Pascual SI. Adv Exp Med Biol. 2009; 652:39-46.
- Discovery, SAR, and Biological Evaluation of a Non-Inhibitory Chaperone for Acid...Discovery, SAR, and Biological Evaluation of a Non-Inhibitory Chaperone for Acid Alpha Glucosidase - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...