

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

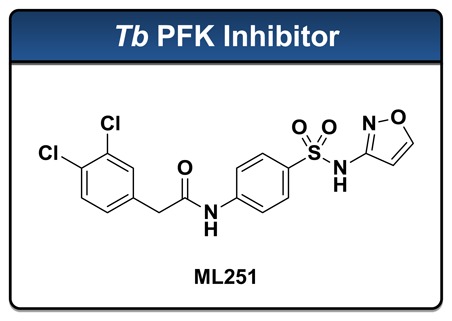
The protist Trypansoma brucei is the causative agent of Human African Trypanosomiasis (HAT), a disease that is endemic to sub-Saharan Africa and is responsible for approximately 50,000 deaths each year worldwide. Current therapies employed to treat HAT generally suffer from poor selectivity profiles, and consequently, lead to high rates of deleterious side effects. Furthermore, growing resistance to these drugs has highlighted a need for the identification of novel therapies. The glycolytic enzyme phosphofructokinase (PFK) has been recognized as a potential therapeutic target in the fight against the bloodstream form of T. brucei due to the reliance of this parasite on the metabolism of glucose as its sole mechanism for the generation of adenosine triphosphates (ATP). The importance of this enzyme has been confirmed by genetic validation, and thus, its inhibition may represent a novel strategy for the treatment of HAT. Currently, no inhibitors of Tb PFK have been described that possess ideal drug-like qualities or submicromolar potency in either enzymatic or live parasite culture assays. As a result, in collaboration with Professor Malcolm Walkinshaw of the University of Edinburgh, a quantitative High Throughput Screening (qHTS) campaign was initiated at the National Institutes of Health (NIH) Chemical Genomics Center (NCGC) to identify novel small molecule inhibitors of Tb PFK. The primary screen utilized a firefly luciferase-coupled assay specifically designed to monitor the production of ADP in the kinase reaction. This work, together with subsequent medicinal chemistry efforts, has identified a novel small molecule inhibitor (ML251) of Tb PFK, which is described herein to possess potent activity in enzymatic assays (IC50 = 370 nM) and toxicity against cultured T. brucei (ED50 = 16.3 μM). This compound has been subjected to a panel of in vitro absorption, distribution, metabolism, and excretion (ADME) assays and delivers a promising profile in this respect. Finally, cytotoxicity evaluations against MRC-5 human lung fibroblast cells and KB-3-1 HeLa cells, in addition to assessment against purified Bacillus stearothermophilus PFK, indicate that ML251 displays a high degree of selectivity for the targeted enzyme.
Assigned Assay Grant #: R03MH092153
Screening Center Name & PI: NIH Chemical Genomics Center, Christopher P. Austin
Chemistry Center Name & PI: NIH Chemical Genomics Center, Christopher P. Austin
Assay Submitter & Institution: Malcolm Walkinshaw, University of Edinburgh
PubChem Summary Bioassay Identifier (AID): 488768
Probe Structure & Characteristics
| CID/ML# | Target Name | IC50/EC50 (nM) [SID, AID] | Anti-target Name(s) | IC50/EC50 (μM) [SID, AID] | Fold Selective | Secondary Assay(s) Name: IC50/EC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 53255421/ML251 | T. bruceiPFK | ADP-Glo: 370 nM IC50 [124391163, 540360] | Cytotoxicity (KB-3-1 Cells) | > 46 μM | > 100 fold | Kinase-Glo: 230 nM IC50 [124391163, 588329] |
Recommendations for Scientific Use of the Probe
The para-amidosulfonamide ML251 represents the first small molecule to possess sub-micromolar inhibitory activity against Trypanosoma brucei phosphofructokinase (Tb PFK). As a molecular probe, ML251 will be invaluable for advancing our understanding of the role that glycolysis plays in the survival of members of the trypanosomatid family. Furthermore, ML251 has shown promising activity in cultured parasite growth assays. Coupled with data suggesting ML251 does not inhibit bacterial isoforms of PFK and lacks cytotoxicity in a number of human cell lines, this result provides the first evidence that specific inhibition of Tb PFK by a small molecule may be realized. Lastly, ML251 has performed extremely well in a myriad of in vitro ADME assessment assays, which provide strong support for ML251 as an excellent lead compound for further development as a small molecule chemotherapeutic for the treatment of T. brucei infection.
1. Introduction
Each year, it is estimated that 50,000 to 70,000 new cases of Human African Trypanosomiasis (HAT), or African Sleeping Sickness, are reported worldwide.[1] This disease is known to be caused via transmission of the parasitic protist Tryanosoma brucei from its insect vector, the tsetse fly, which inhabits much of mid-continental Africa. This region is home to roughly 60 million people, all of whom remain at risk for this disease. The onset of HAT is characterized by significant swelling within the lymphatic system, especially in the nodes at the base of the neck (Winterbottom’s sign). Following this haemolymphatic stage of the disease, a neurological stage begins, in which the parasite invades the central nervous system via penetration of the blood-brain barrier. It is during this stage that the disease earns its nickname “sleeping sickness,” as common symptoms include confusion, reduced coordination, and a disrupted sleep cycle. Three subspecies of T. brucei are known, all of which contribute differently to the progression of HAT. Infection by T. b. gambiense results in slow onset of chronic trypanosomiasis, while T. b. rhodesiense gives rise to fast onset of the acute form of the disease. The third subspecies, T. b. brucei, is not human infective, but is associated with Nagana, or animal African trypanosomiasis. Like other diseases caused by members of the trypanosomatid family (e.g. Chagas’ disease and leishmaniasis), HAT is invariably fatal without proper medical care. Unfortunately, treatment is often complicated by issues associated with drugs that are currently employed to treat this disease (Table 1).[1] Furthermore, an emerging drug resistance within T. brucei highlights a pressing need for the development of new compounds that are active against novel therapeutic targets.
Table 1
Drugs available for the treatment of trypanosomatid-borne diseases.
The catabolic processing of glucose to pyruvate has been demonstrated to represent the only viable pathway for the production of ATP in the bloodstream form of T. brucei.[2] Furthermore, mitochondrial oxidative phosphorylation processes are known to be repressed in these parasites. Therefore, glycolysis as a whole represents an attractive target for emerging chemotherapeutics.[2] This is supported by RNA interference (RNAi) experiments that indicate that only partial decreases in overall glycolytic flux are necessary in order to significantly reduce survival rates of cultured parasites.[3]
With regard to specific glycolytic enzymes, phosphofructokinase (PFK) represents an attractive target for the development of selective small molecule inhibitors, as its activity has been genetically validated in T. brucei.[3] This enzyme facilitates the conversion of fructose 6-phosphate (F6P) to fructose 1,6-bisphosphate (F1,6BP) and utilizes ATP as a phospho-donor in an essentially irreversible transformation. In addition, Tb PFK has been well characterized with respect to crystallographic structural analyses,[4] is available in high yield via bacterial expression systems,[5] and may be monitored via sensitive and convenient enzyme-activity assays.[6] Finally, elegant structural and kinetic studies have demonstrated that Tb PFK possesses a high degree of inimitability when compared to functional homologues that are expressed in other species, including humans.[7] Combined, this information suggests potential for the discovery of a novel, effective, and specific small molecule inhibitor of Tb PFK.
Presently, a diminutive number of non-specific small molecule inhibitors of Tb PFK have been described in the literature (Figure 1), each of which is required in micromolar quantities to impart inhibitory activity.[8–10] It is noteworthy that to date, no high-throughput screening campaigns have been reported in which Tb PFK has directly been targeted.
Figure 1
Known small molecule inhibitors of Tb PFK.
2. Materials and Methods
General Methods for Chemistry
All air or moisture sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Anhydrous solvents such as dichloromethane, N,N-dimethylforamide (DMF), acetonitrile, methanol and triethylamine were purchased from Sigma-Aldrich. Preparative purification was performed on a Waters semi-preparative HPLC system. The column used was a Phenomenex Luna C18 (5 micron, 30 × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10% to 50% acetonitrile over 8 minutes was used during the purification. Fraction collection was triggered by UV detection (220 nM). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA). Method 1: A 7 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 minute run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 micron, 3 × 75 mm) was used at a temperature of 50 °C. Method 2: A 3 minute gradient of 4% to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 minute run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 micron, 3 × 100 mm) was used at a temperature of 50 °C. Purity determination was performed using an Agilent Diode Array Detector for both Method 1 and Method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical Shifts are reported in ppm with undeuterated solvent (DMSO-h6 at 2.49 ppm) as internal standard for DMSO-d6 solutions. All of the analogs tested in the biological assays have a purity greater than 95% based on both analytical methods. High resolution mass spectrometry was recorded on Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formulae was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
2.1. Assays
2.1.1. Tb PFK qHTS assay
Inhibition of T. brucei PFK was screened by coupling PFK-mediated ADP production to a modified luciferase-based detection assay (ADP-Glo, Promega, Madison, WI). Though the exact contents of the detection reagent are proprietary, in essence it utilizes coupled enzyme reactions to 1) deplete all unconverted ATP remaining from the initial PFK reaction, 2) reconvert all PFK-hydrolyzed ADP back to ATP and 3) produce a luminescent signal using the ATP-dependent luciferase reaction. Thus, a decrease in luminescence is indicative of PFK inhibition. As no PFK-specific inhibitors have been previously identified, an enzyme-free control was included on each plate for 100% inhibition data normalization. A stepwise description of the 1536-well assay is shown in Table 2.
Table 2
ADP-Glo Tb PFK qHTS assay protocol.
2.1.2. Tb PFK ATP-depletion secondary assay
Confirmed qHTS active compounds were tested in a modified qHTS assay using identical enzyme and substrate conditions to the primary assay, yet with an alternative measure of PFK activity. The primary qHTS assay provides a luminescence-based detection of ADP production, and thus a direct measurement of Tb PFK product formation. An alternative detection kit, Kinase-Glo Plus (Promega, Madison, WI), utilizes a comparable luminescence-based technology to measure ATP concentration. When used in conjunction with optimized Tb PFK conditions, this orthogonal assay provides a measurement of PFK activity as a function of ATP depletion. Upon inhibition of PFK, luminescent signal remains high relative to uninhibited enzyme, providing a change in signal opposite to that seen in the primary assay. This allows for both confirmation of inhibitor activity, as well as a controlled counter-screen to those compounds that act on luciferase or otherwise inhibit the detection reagent. Both kits detect final nucleotide levels with similar coupling enzymes, and therefore compounds that directly inhibit the detection step should lead to a reduction in luminescent signal in both ADP and ATP assays, whereas true inhibitors should lead to opposing changes in luminescence, generally with comparable potency (see Figure 7). A stepwise description of the 1536-well assay is shown in Table 3. A variation of this protocol was also developed to preliminarily assess the mode of inhibition for identified inhibitors; either ATP or F6P was included at a saturating concentration (1 mM and 5 mM, resp.) with all remaining reagent concentrations held constant. Inhibitor potencies were then assessed to determine if ML251 activity was differentially reduced with inclusion of excess substrate.
Table 3
Tb PFK Kinase-Glo assay protocol.
2.1.3. Human cell line toxicity counterscreen
Confirmed qHTS active compounds were tested for toxicity against the MRC-5 human lung fibroblast and KB-3-1 human cervical carcinoma cell lines. These lines have historically been used as surrogate hosts in in vitro Trypanosoma assays, and so provide a relevant surrogate for human cells in testing Tb PFK inhibitor toxicity. Briefly, MRC-5 and KB-3-1 cultured cells were plated in phenol red-free Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) at 1,000 cells per well, dosed with titrated compounds in DMSO and incubated for 72 hours at 37 °C. Toxicity was assessed using the CellTiter Glo kit (Promega, Madison, WI), which uses intracellular ATP concentration as a viability marker. As inhibitors of glycolysis could have potentially affected total cellular ATP levels, an ATP-independent viable dye protocol was prepared (MTT, Invitrogen, Carlsbad, CA), though it was not required, as none of the active compounds demonstrated appreciable toxicity in the CellTiter Glo assay.
2.1.4. Tb PFK substrate competition assay
The mechanism of Tb PFK inhibition was additionally tested for select compounds, using substrate competition to examine whether compounds competed with ATP for F6P for binding. Competition was assayed by coupling production of ADP by Tb PFK to rabbit pyruvate kinase (PyK), which converts ADP and phosphoenol pyruvate (PEP) to ATP and pyruvate. The reaction is further coupled to lactate dehydrogenase (LDH), which converts pyruvate and NADH to lactate and NAD+, respectively. NADH conversion can be kinetically measured by a spectrophotometer using absorbance at 340 nm. Briefly, a master substrate mix consisting of 25 nM Tb PFK, saturating concentrations of one substrate (either ATP or F6P), excess rabbit PyK, PEP and LDH, and 0.18 mM NADH was prepared. Titrations of the competing substrate (either ATP or F6P) and small molecule inhibitor spanning their respective Kms and IC50s were also prepared. Individual aliquots of substrate mix, competing substrate and inhibitor, were then mixed to begin the reaction, which was immediately transferred to a quartz cuvette and read on a Varian spectrophotometer; kinetic change in absorbance was measured at 340 nm for 20 seconds. The changes in absorbance were plotted for each pair of substrate and inhibitor concentrations to determine initial velocities, which were then transformed to Lineweaver-Burk plots for demonstration of binding site competition.
2.1.5. Tb PFK thermal melt assay
A fluorescence-based thermal denaturation assay was performed in a BioRad iQ5 rtPCR Thermocycler. Each sample contained 0.1 mg/mL of Tb PFK, 5× SYPRO orange (dilution 1/1000 from stock Sigma S5692), and 0.5 mM of ligand (F1,6BP, F6P, ATP or AMP) in 50 μL of 1× buffer (10 mM MgCl2, 100 mM KCl, 100 mM TEA, 10% glycerol, pH 7.2). Inhibitors were tested with a final concentration of 50 μM. The 96-well plate containing all of the samples was heated at a rate of 1.0 °C/min from 20 to 80 °C, and the fluorescence intensity was measured with excitation/emission wavelengths of 490 and 580 nm, respectively. Tm values were provided by the software CFX Manager. Each sample was tested in duplicate.
2.1.6. T. brucei in vitro parasite growth assay
Selected active compounds were additionally tested in a well-validated in vitro parasite assay to determine the relative potencies of lead compounds against live T. brucei cultures. Briefly, dilutions of T. brucei brucei were incubated in modified Iscove’s cell culture medium (supplemented with 10% FBS, 0.05 mM bathocuproine sulfonate, 1.5 mM L-cysteine, 1 mM hypoxanthine, 0.2 mM 2-mercaptoethanol, 1 mM sodium pyruvate and 0.16 mM thymidine) with titrations of compound for 72 hours. The cell density is adapted so that after 72 hours of incubation, the cells are at the end of the logarithmic growth phase (~5×104 tryps/mL). General morphology and motility were recorded after incubation, and Alamar Blue dye was added as a fluorescent marker of viability (ex530/em590) and read on a fluorescent plate reader following 4 hrs of incubation at 37 °C. The results of this assay were used to prioritize medicinal chemistry efforts in conjunction with the human cell toxicity assay (see Section 2.1.3) to give a measure of relative parasite/host toxicity ratio.
2.2. Probe Chemical Characterization

Probe Characterization of ML251
*Purity >95% as determined by LC/MS and 1H NMR analyses.
2-(3,4-dichlorophenyl)-N-(4-(N-isoxazol-3-ylsulfamoyl)phenyl)acetamide: LC-MS Retention Time: t1 (Method 1) = 5.734 min and t2 (Method 2) = 3.624 min; 1H NMR (400 MHz, DMSO-d6) δ ppm 11.45 (1 H, br. s.), 10.60 (1 H, s), 8.70 (1 H, d, J=1.4 Hz), 7.78 – 7.83 (2 H, m), 7.72 – 7.78 (2 H, m), 7.55 – 7.62 (2 H, m), 7.30 (1 H, dd, J=8.3, 1.7 Hz), 6.40 (1 H, d, J=1.6 Hz), 3.73 (2 H, s); HRMS (ESI) m/z (M+H)+ calcd. for C17H14Cl2N3O4S, 426.0077; found 426.0077.
MLS numbers for probe analogs
| Internal ID | MLS ID | SID | CID | ML # | Type | Source |
|---|---|---|---|---|---|---|
| NCGC00244858 | MLS003678599 | 124391163 | 53255421 | ML251 | Probe | NCGC |
| NCGC00244099 | MLS003678600 | 124391216 | 53255380 | Analog | NCGC | |
| NCGC00244854 | MLS003678601 | 124391196 | 5220784 | Analog | NCGC | |
| NCGC00244881 | MLS003678602 | 124391164 | 2434603 | Analog | NCGC | |
| NCGC00244866 | MLS003678603 | 124391149 | 53255430 | Analog | NCGC | |
| NCGC00248353 | MLS003678604 | 124391211 | 53255399 | Analog | NCGC |

Figure 2List and structures of probe and analogs that have been submitted to the MLSMR
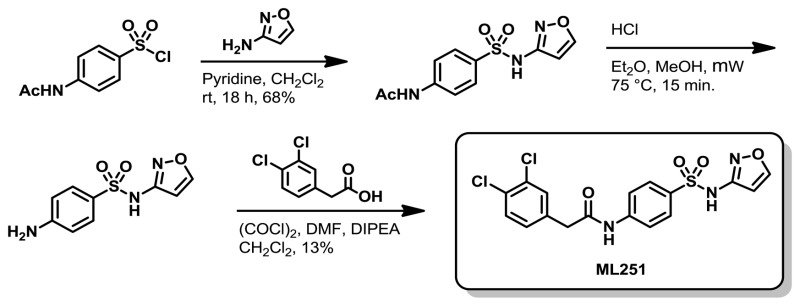
Scheme 1Synthetic route to ML251
2.3. Probe Preparation
Preparation of 2-(3,4-dichlorophenyl)-N-(4-(N-isoxazol-3-ylsulfamoyl)phenyl)acetamide (ML251)
Step 1: Pyridine (0.208 mL, 2.57 mmol) and 4-acetamidobenzene-1-sulfonyl chloride (0.500 g, 2.14 mmol) were added sequentially to a solution of isoxazol-3-amine (0.198 g, 2.35 mmol) in CH2Cl2 (21.4 mL) were added. The reaction was capped and allowed to stir at ambient temperature overnight. Following concentration in vacuo, the crude residue was purified via silica gel chromatography using a 95:5 – 0:100, hexane/EtOAc (v/v) gradient to give N-(4-(N-isoxazol-3-ylsulfamoyl)phenyl)acetamide as a viscous oil (413 mg, 68%).
Step 2: A 2.0 M solution of HCl in Et2O (0.800 mL, 1.60 mmol) was added to a solution of N-(4-(N-isoxazol-3-ylsulfamoyl)phenyl)acetamide (0.225 g, 0.80 mmol) in MeOH (8.0 mL). The reaction vial was capped and heated to 75 °C for 15 min in a microwave reactor. After cooling, the reaction was concentrated in vacuo to give crude 4-amino-N-(isoxazol-3-yl) benzenesulfonamide, HCl as a white solid that was judged to be >95% pure by LC/MS analysis. This material was therefore used in subsequent reactions without further purification.
Step 3: Oxalyl chloride (0.092 mL, 1.05 mmol) and DMF (5.8 μL, 0.08 mmol) were added to a solution of 2-(3,4-dichlorophenyl)acetic acid (0.154 g, 0.75 mmol) in CH2Cl2 (1.25 mL), which resulted in vigorous evolution of gas. The reaction was left to stir for 2.5 hr and was then concentrated in vacuo. The residue was then taken up in CH2Cl2 (1.25 mL) and added to a solution of 4-amino-N-(isoxazol-3-yl)benzenesulfonamide (0.090 g, 0.38 mmol) and DIPEA (0.197 mL, 1.13 mmol) in CH2Cl2 (1.25 mL) at ambient temperature. After 30 min at the same temperature, the reaction was concentrated in vacuo and the residue was taken up in EtOAc (15 mL), washed with water (2 × 15 mL) and brine (2 × 15 mL), and dried over anhydrous Na2SO4. The mixture was filtered and concentrated in vacuo to give an orange oil that was taken up in 1.5 mL DMSO and purified via reverse phase chromatography to give 2-(3,4-dichlorophenyl)-N-(4-(N-isoxazol-3-ylsulfamoyl)phenyl)acetamide, TFA (ML251) as a white solid (21 mg, 13%).
3. Results
3.1. Summary of Screening Results
To identify small molecule inhibitors of Tb PFK, a robust luminescence-based assay was developed and miniaturized to 1536-well format. Prior to the full-collection screen, the assay was tested and found to perform reproducibly by screening the Library of Pharmacologically Active Compounds (LOPAC1280) in triplicate using a fully-integrated robotic system. The assay was then screened against the 330,686-compound MLPCN library arrayed as six-point titrations ranging from 57 μM to 3.7 nM (AID: 485367).
In total, 1,761 assay plates were screened in one continuous robotic run; 251 plates (14% of the full-scale screening plates) failed due to mechanical reasons (usually the pin tool and dispensing problems) and were identified for re-screening. These plates were added at the end of the run and the failed plates were not included for calculations. Otherwise, the assay performed well during the remaining course of the screen with the Z′ factor remaining consistent with an average value of 0.75 (Figure 4A). In addition, the percent coefficient of variation among wells was consistently low, with average value of 6.9% (Figure 4B).

In contrast to traditional HTS, qHTS provides concentration response curves (CRCs) for each compound and allows for calculation of an IC50 value (defined as the half-maximal activity concentration) for each compound in the primary screen. In this screen, the inhibition associated with each well was computed from the endpoint luminescence and normalized against control wells. Analysis of the CRCs (curve quality and potency) resulted in 1,054 inhibitors with single or dual asymptote CRCs and greater than 50% efficacy (class -1.1 and -1.2, Figure 4C) and 242 activators (class 1.1, 1.2, 2.1 and 2.2, Figure 4C), representing 0.31% and 0.07% of the chemical library, respectively. Compounds were further filtered by using the qHTS Assay for Inhibitors of Firefly Luciferase (AID 588342) as a counterscreen to identify those with the potential to directly inhibit luciferase in the detection reagent. However, as luciferase and Tb PFK share a common substrate in ATP, compounds capable of competing with ATP binding could conceivably bind to and inhibit both enzymes. As such, we only excluded compounds whose potencies were comparable in both Tb PFK and purified luciferase assays, suggesting that the perceived inhibition was primarily governed by competition with luciferase itself.
The 1,056 compounds found to be free of luciferase inhibitory activity were tested in the ATP depletion secondary assay for orthogonal confirmation, as well as the human cell line toxicity assay for toxicity assessment, resulting in the identification of the present chemotype. The initial active from the probe chemotype was MLS002158384-01/CID 2431573. Subsequent analogs of the probe chemotype were tested in the primary PFK ADP-Glo assay and human cell toxicity counterscreen, with select analogs being additionally tested in the PFK substrate competition, thermal melt and in vitro parasite toxicity assays. A summary of the work-flow associated with this project is given in Figure 5. These efforts resulted in the identification of a compound, CID 2431573, that possessed desirable activity profiles and in vitro metabolic properties, in addition to an attractive chemical scaffold for the development of analogs as a means to establish SAR (Figure 6).

Figure 5
Work flow for the identification of small molecule inhibitors of Tb PFK. (Note: # of compounds in this flowchart indicates the number of compounds that passed the filters)

Figure 6
Structure and selected physical properties of the qHTS hit CID-2431573 and its performance in the 1° and 2° assays.
3.2. Dose Response Curves for Probe

Figure 7Graphical representation of the percent inhibition of Tb PFK by ML251
Results shown using both the primary ADP production assay (black circles) and the Kinase-Glo ATP depletion assay (blue circles).
3.3. Scaffold/Moiety Chemical Liabilities
In assessing potential liabilities of ML251, there were three major functionalities that were considered. The first was the amide bond and its potential to be cleaved/hydrolyzed to give a carboxylic acid and an aniline, which could present genotoxicity problems. Second, the presence of the un-substituted benzylic methylene functionality was initially viewed as a potential liability for microsomal oxidation. Third, the isoxazole group presents the possibility of cleavage and scission reactions in liver microsomes. It should be noted that the probe contains the core of sulfamethoxazole, which is a bacteriostatic antibiotic used in vivo. So, while these general concerns over liability are warranted and worth considering, there is evidence that a very close analog containing two of the three functionalities discussed (aniline and isoxazole) has appropriate PK/PD properties to warrant in vivo use. It should also be noted that sulfa drugs have liabilities for causing Stevens-Johnson Syndrome, although the majority of these drugs have either a primary sulfonamide or free aniline functionalities. The remaining liability, the benzylic methylene group, remains as an issue of which to be aware, but it is also present in numerous drugs and agents used in vivo. Importantly, in vitro assessment of stability of ML251 against mouse and human microsomes, human plasma (Table 7), and sustained stability in DPBS and ADP-Glo assay buffers (Figure 3), prove that the probe possesses excellent overall stability.

Figure 3
Stability of ML251 in ADP-Glo assay buffer (Panel A) and DPBS (pH 7.4) buffer (Panel B) at room temperature over 48 hr.
3.4. SAR Tables
Table 4Tb PFK Inhibition: Representative Analogs
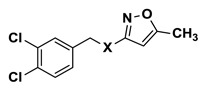 | |||||
|---|---|---|---|---|---|
| Entry | Internal IDa | SID | CID | X | IC50 (μM)b |
| 1 | NCGC00242251-01 | 124391134 | 2431573 |
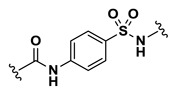 | 0.41 |
| 2 | NCGC00244111-01 | 124391165 | 53255402 |
 | 16.34 |
| 3 | NCGC00244098-01 | 124391168 | 53255386 |
 | 5.80 |
| 4 | NCGC00244099-01 | 124391216 | 53255380 |
 | 32.61 |
| 5 | NCGC00244110-01 | 124391180 | 53255442 |
 | 32.61 |
| 6 | NCGC00244106-01 | 124391166 | 53255422 |
 | Inactive |
| 7 | NCGC00244097-01 | 124391175 | 53255427 |
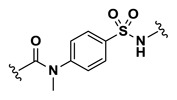 | 1.63 |
| 8 | NCGC00244100-01 | 124391176 | 53255426 |
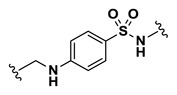 | 4.61 |
| 9 | NCGC00244105-01 | 124391179 | 53255440 |
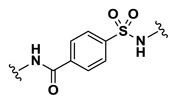 | 2.91 |
| 10 | NCGC00244107-01 | 124391170 | 53255435 |
 | 36.59 |
| 11 | NCGC00244096-01 | 124391174 | 53255438 |
 | 2.91 |
| 12 | NCGC00244108-01 | 124391172 | 53255391 |
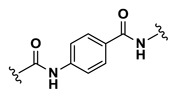 | Inactive |
| 13 | NCGC00244104-01 | 124391178 | 53255404 |
 | 2.59 |
- a
All compounds synthesized at NCGC.
- b
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the ADP-Glo assay.
Table 5Tb PFK Inhibition: Representative Analogs
 | |||||
|---|---|---|---|---|---|
| Entry | Internal ID | SID | CID | X | IC50 (μM)a |
| 14 | NCGC00244854-01 | 124391196 | 5220784 |
 | 36.59 |
| 15 | NCGC00244859-01 | 124391227 | 53255378 |
 | 7.30 |
| 16 | NCGC00244879-01 | 124391215 | 1290388 |
 | 1.83 |
| 17 | NCGC00244872-01 | 124391191 | 53255419 |
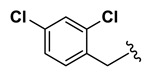 | 4.61 |
| 18 | NCGC00244865-01 | 124391190 | 53255387 |
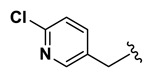 | 20.57 |
| 19 | NCGC00244869-01 | 124391152 | 53255383 |
 | 0.46 |
| 20 | NCGC00244866-01 | 124391149 | 53255430 |
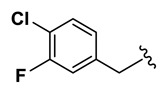 | 0.58 |
| 21 | NCGC00244868-01 | 124391151 | 53255432 |
 | 5.80 |
| 22 | NCGC00244863-01 | 124391154 | 53255434 |
 | 18.34 |
| 23 | NCGC00244861-01 | 124391201 | 53255406 |
 | Inactive |
| 24 | NCGC00244881-01 | 124391164 | 2434603 |
 | Inactive |
| 25 | NCGC00244860-01 | 124391200 | 53255376 |
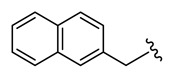 | 6.51 |
| 26 | NCGC00244853-01 | 124391226 | 1010711 |
 | 14.56 |
| 27 | NCGC00244856-01 | 124391195 | 53255385 |
 | 5.17 |
| 28 | NCGC00244877-01 | 124391158 | 53255423 |
 | 36.59 |
- a
All compounds synthesized at NCGC.
- b
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the ADP-Glo assay.
Table 6Tb PFK Inhibition: Representative Analogs
 | |||||
|---|---|---|---|---|---|
| Entry | Internal ID | SID | CID | X | IC50 (μM)a |
| 29 | NCGC00248360-01 | 124391204 | 53255437 |
 | 0.65 |
| 30 ML251 | NCGC00244858-01 | 124391163 | 53255421 |
 | 0.37 |
| 31 | NCGC00248353-01 | 124391211 | 53255399 |
 | 0.21 |
| 32 | NCGC00248350-01 | 124391225 | 53255393 |
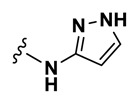 | 0.52 |
| 33 | NCGC00248341-01 | 124391234 | 53255401 |
 | 1.46 |
| 34 | NCGC00248366-01 | 124391222 | 22829926 |
 | 16.34 |
| 35 | NCGC00248361-01 | 124391219 | 53255416 |
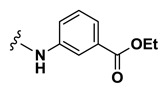 | Inactive |
| 36 | NCGC00248362-01 | 124391220 | 53255418 |
 | 0.26 |
| 37 | NCGC00248345-01 | 124391223 | 53255425 |
 | 0.16 |
| 38 | NCGC00248352-01 | 124391232 | 53255407 |
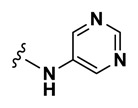 | 0.82 |
| 39 | NCGC00248342-01 | 124391207 | 53255382 |
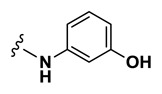 | 1.46 |
| 40 | NCGC00248343-01 | 124391208 | 53255377 |
 | 5.80 |
- a
All compounds synthesized at NCGC.
- b
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the ADP-Glo assay.
3.5. Cellular Activity
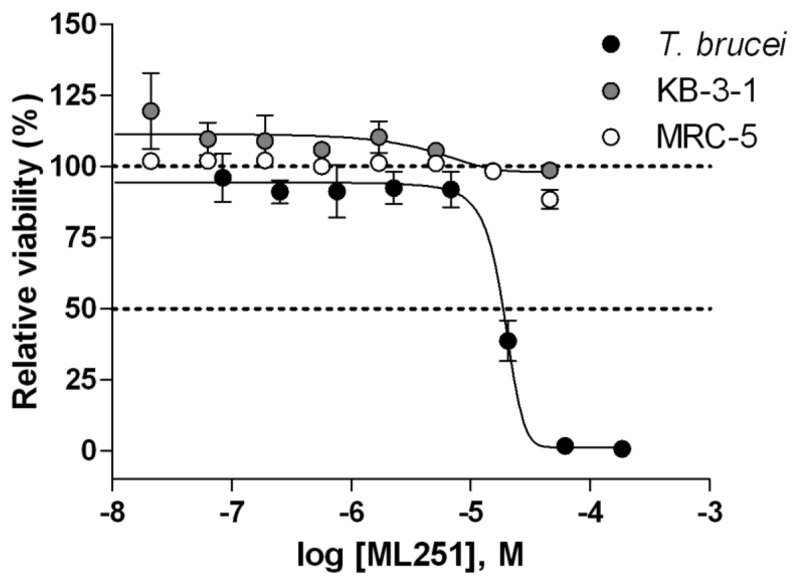
Figure 8Cytotoxicity of ML251
Against cultured T. brucei brucei (black circles), KB-3-1 human cervical carcinoma (grey circles) and MRC-5 human lung fibroblast (white circles) cell lines after 72 hr incubation. Each point represents the mean±SD for three independent experiments.

Figure 9Growth inhibitory activity of selected analogs against cultured T. brucei brucei after 72 hr incubation
Each point represents the mean±SD for four independent experiments.
3.6. Profiling Assays

Figure 10Inhibitory activity of ML251
Against T. cruzi (grey circles), T. brucei (black circles), and B. stearothermophilus (white circles) isoforms of PFK.
Table 7ADME profile for qHTS hit 1 (CID-2431573) and 30 (ML251)
| Compound | Aqueous Kinetic Solubility | Liver Microsomal
Stability (t1/2 in min.) | Plasma
Stability (% remaining after 2 h) | |||
|---|---|---|---|---|---|---|
| μg/mL | μM | Mouse | Human | Mouse | Human | |
| CID-2431573 | 23.3 | 45.0 | 408 | N/D | N/D | N/D |
| ML251 | > 81.0 | > 150.0 | 231 | 330 | 100 | 100 |
4. Discussion
Efforts to establish tractable SAR around the screening hit MLS002158384 (CID 2431573) began with its resynthesis to confirm activity. In doing so, 1 (Table 4, CID 2431573) was identified to have an inhibitory potency of 410 nM against Tb PFK in the ADP-Glo assay. Initial rounds of analog synthesis focused on changes to the para-amidosulfonamide core of the molecule (Table 4). These studies revealed 1,4-disubstitution about the phenyl ring to be optimal as meta-substituted analog 2 (CID 53255402) suffered from greatly diminished activity. In addition, installation of extended aromatic (3, CID 53255386), saturated (4, CID 53255380), alkyl chain (5, CID 53255442), and heteroaryl (6, CID 53255422) cores all led to less active derivatives. Initial rounds of synthesis concluded with efforts to understand the importance of the amide and sulfonamide linkers. These studies indicated very tight SAR with respect to both of these functionalities, as indicated in analogs 7 – 13.
The 3,4-dichlorobenzyl motif of 1 was hypothesized to be of significant importance to overall activity within the series due to its presence in a number of hits identified in the qHTS campaign. This theory was supported by the trends observed in the analogs listed in Table 5. Single- and double-point deletions of the chloro groups led to less active compounds (14 – 16). However, from these results, it was recognized that chloro-substitution at the 4-position of this ring generally imparted greater control on overall activity than at the 3-position. This finding was supported in compounds 19 – 20, which only suffered from marginal decrease in activity, despite replacement of the 3-chloro functionality with varying electron-withdrawing, hydrophobic moieties. Further manipulations of this region of the molecule, in the form of electon-donating lipophilic and hydrophilic functionalities, led to diminished activity (21 – 25). Finally, the importance of the benzylic methylene group was highlighted via reduced potencies observed in phenyl (26, CID 1010711), homo-benzyl (27, CID 53255385), and dimethylbenzyl (28, CID 53255423) analogs. Of particular note from this round of synthesis is analog 24 (CID 2434603) which bears a 3,4-dimethoxybenzyl substitution. This compound showed a complete lack of activity in the ADP-Glo assay and proved to have excellent aqueous solubility (> 150 μM), as determined in in vitro kinetic solubility assessments. Therefore, analog 24 was identified as an inactive control for the project and was utilized in this capacity for all subsequent studies.
While a number of analogs had been generated in the first two rounds of synthesis that possessed comparable activity to the original hit 1, failure to improve upon this potency led to the decision to utilize the previously solved ATP-bound crystal structure of Tb PFK [7] as a basis for molecular modeling efforts to guide future rounds of analog development. Thermal melt data suggested that 1 engages in competition for binding with F6P, as stabilization of Tb PFK is observed in the presence of ATP and inhibitor, but not in the presence of F6P and inhibitor (data not shown). This finding led to docking of 1 in the sugar binding site (Figure 11, Panel A) and conformational energy minimization delivered a binding orientation in which the 3,4-dichlorobenzyl functionality of 1 extends into a hydrophobic pocket generated by the Leu346, Ile347, and Ile349 residues of Tb PFK (Figure 11, Panel B). Important hydrogen bonding interactions are also predicted in this rendering for the sulfonamide linker with the Arg383 residue (Figure 11, Panel C). Lastly, the modeling predicts occupation of the 5-methyl-3-aminoisoxazole functionality within a highly hydrophilic region of the Tb PFK active site. It is within this site that the LYS226 residue is predicted to engage in a hydrogen bond with the nitrogen center of the isoxazole functionality of 1. This suggested mode of binding was further corroborated by mechanistic enzyme studies that were performed on ML251. Inclusion of saturating concentrations of ATP or F6P in the ADP-Glo inhibition assay showed a marked right-shift in ML251 potency under high F6P conditions (Figure 13, Panel A); the IC50 of ML251 against Tb PFK shifted from 365 nM under standard conditions to 2.06 μM in the presence of saturating F6P, demonstrating a 5.6-fold shift in potency. Inclusion of saturating ATP, by contrast, demonstrated virtually no change in ML251 potency, with an IC50 of 326 nM. These observations were followed up with a mechanistic competition study in which individual substrates and ML251 were titrated in a coupled PFK assay system. ML251 demonstrated a minimal effect on the Km of ATP, yet appeared to affect Vmax (Figure 13, Panel B, transformed to Lineweaver-Burk plot for ease of visualization), suggesting that it is noncompetitive with respect to ATP. F6P, conversely, showed no change in Vmax in the presence of ML251, but did show a shift in Km, suggesting that the compound may be competitive with respect to F6P (Figure 13, Panel C). These observations agree with the results of both our biochemical assays and molecular modeling in suggesting that the lead chemical series preferentially binds in the F6P region of the Tb PFK active site.

In accordance with these findings, a third round of analog synthesis was undertaken to explore beliefs that appending hydrophilic, anionic functionalities to the isoxazole moiety would best facilitate binding within the Tb PFK active site. These results are listed in Table 6 and do confirm these theories very well. In an effort to decrease the lipophilicity in this region of the molecule, analog 30 (Table 6, ML251) was synthesized, in which the methyl group at the 5-position of the isoxazole was removed. This change yielded the first analog to show improved potency relative to 1. In addition, compound 31 (CID 53255399), bearing a reversed isoxazole functionality relative to 1, also showed improved activity in the ADP-Glo assay. All additional analogs bearing various five-membered heterocycles (e.g. 32 (CID 53255393) and 33 (CID 53255401)) possessed decreased potency against Tb PFK. A phenyl ring scan was performed and provided remarkable SAR with respect to the value of negatively charged functionalities within this portion of the molecule. Both the phenyl (34, CID 22829926) and meta-ethylbenzoate (35, CID 53255416) derivatives showed significant loss of activity in the ADP-Glo assay. However, simple hydrolysis of 35 to the corresponding benzoic acid derivative 36 (CID 53255418) gave rise to a highly potent analog. The corresponding para-substituted benzoic acid 37 (CID 53255425) also retained excellent activity. Clearly, installation of a benzoic acid functionality presents liabilities when eventual in vivo translation is desired. However, this result provides excellent evidence supporting the predicted binding mode illustrated in Figure 11. A complete summary of the SAR generated from evaluation of analogs in the ADP-Glo assay is offered in Figure 12.
Figure 12
SAR Summary.
In an effort to probe the selectivity of the series for Tb PFK, all analogs were evaluated for inhibitory activity against the PFK isoform of Trypanosoma cruzi, a related trypanosomatid species responsible for Chagas disease, as well as that of Bacillus stearothermophilus, a thermophilic species of bacteria. Not surprisingly, comparable inhibitory activity against T. brucei PFK and T. cruzi PFK was observed for all compounds, as these isoforms display a high degree of similarity, with greater than 90% sequence identity.[11] Cross-species activity of small molecules across multiple members of the trypanosomatid family is a well-recognized phenomenon, and given the tremendous burden both T. brucei and T. cruzi place on global health, this shared inhibition of PFK would not be considered a liability for the lead series.[12,13] Encouragingly, when the same compounds were tested against the B. stearothermophilus isoform of PFK, no inhibitory activity was observed (PFK isoform selectivity data for ML251 given in Figure 10). As the Bacillus isoform shares only 30% sequence identity with T. brucei, this lack of inhibitory activity seems relatively unsurprising.[14] Following these studies, all analogs were evaluated for general cytotoxicity against MRC-5 human lung fibroblast and KB-3-1 human cervical carcinoma cell lines, as purified human PFK is not available. These studies revealed essentially no cytotoxicity for the lead series, providing further evidence of its selectivity for trypanosomatid isoforms of PFK (cytotoxicity data for ML251 is given in Figure 8).
With a number of submicromolar inhibitors of Tb PFK in hand, a collaborator was identified to assess the ability of a small number of analogs to inhibit the growth of cultured T. brucei brucei. This subspecies, while not human infective, is highly related to both T. b. gambiense and T. b. rhodesiense and is commonly used as a model in laboratory settings. Initial evaluation of the original screening hit (1, ED50 = 26.8 μM) and inactive control (24, ED50 > 200 μM) in this assay provided encouraging results (Figure 9). However, analogs 36 (ED50 = 105.2 μM) and 37 (ED50 > 200 μM), which were among the most potent inhibitors of Tb PFK, proved highly ineffective against cultured T. brucei brucei. Furthermore, analog 31 (ED50 = 157 μM), bearing a reversed isoxazole functionality, also displayed poor inhibitory activity in these experiments despite possessing high potency in enzymatic assays. With regards to the dichlorobenzyl functionality of 1, the performance of the 4-chloro-3-fluorobenzyl analog 20 (ED50 = 38.9 μM) provided evidence that changes to this region of the molecule are detrimental to overall activity of the series against cultured parasites. Ultimately, the 5-aminoisoxazole derivative 30 (ML251, ED50 = 16.3 μM) proved to be the only analog to display improved potency relative to 1 in these studies. It should be noted that the overall reduction in potency relative to the ADP-Glo assay was anticipated, as a myriad of studies have shown translation from enzymatic to cultured parasite assays is often accompanied by significant loss of activity[15,16]. However, in establishing viable activity in these in vitro parasite growth assays, the decision was made to declare ML251 as an optimized small molecule inhibitor of Tb PFK. Furthermore, the lack of cytotoxicity displayed by ML251 against various human cell lines (Figure 8), coupled with a promising ADME profile (Table 7) and selectivity for PFK isoforms within the trypanosomatid family (Figure 10), support its consideration as a lead compound for further development to ultimately validate Tb PFK as a therapeutic target.
4.1. Comparison to Existing Art and How the New Probe is an Improvement
Prior to our efforts, only a very small number of non-specific inhibitors of Tb PFK had been identified through targeted screening approaches (Table 8). These compounds generally lack ideal drug-like qualities and possess significant molecular complexity. In the case of Suramin,[8] substantial molecular weight and multiple sulfonic acid functionalities account for its poor PK profile. With regards to Polysin[9] and the glycoside-based inhibitor described by Nowicki and co-workers,[10] stereochemical complexity inherent to each ultimately render these molecules as poor candidates for further development via medicinal chemistry optimization. All of these issues are ameliorated in ML251, which is the first Tb PFK inhibitor to display submicromolar potency. PFK’s role in catalyzing the first irreversible step in glycolysis lends it considerable control over the flux of the entire pathway. As such, this probe should provide the research community with a useful tool to 1) further elucidate PFK’s influence on glycolysis and energy production in trypanosomes and 2) potentially validate glycolytic enzymes as viable drug targets in the treatment of trypanosomatid-related diseases. Notably, favorable physical and in vitro metabolic properties, specificity within the trypanosomatid family, and activity in in vitro cultured parasite assays suggest that ML251 will inevitably prove to be an improvement over the described prior art.
Table 8
Comparison of probe to prior art.[17]. ML251 demonstrates a better potency against Tb PFK when compared to the approved trypanosomiasis drug suramin[8] and the known PFK inhibitors polysin[9] and a glycoside analog[10].
4.2. Mechanism of Action Studies
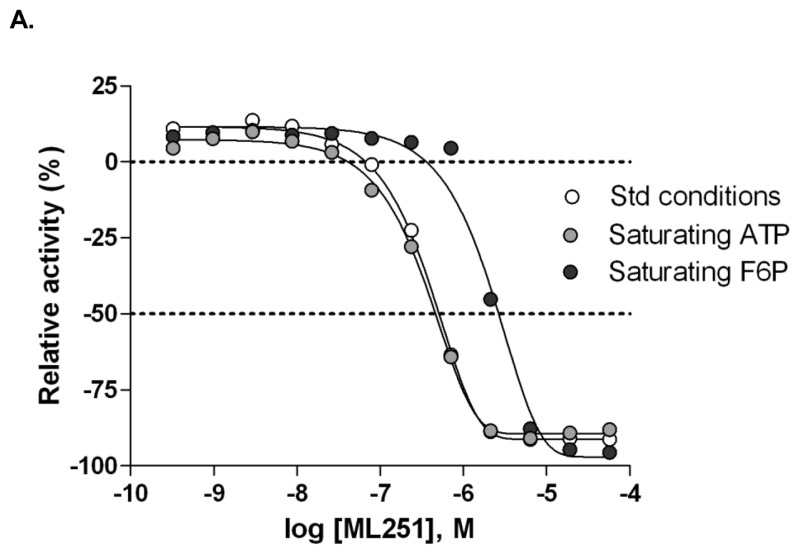

Figure 13Evaluation of substrate competition of ML251 with F6P and ATP in Tb PFK
A. Inhibitory activity of ML251 in ADP-Glo assay under standard (white circles), saturating ATP (light grey circles) and saturating F6P (dark grey circles) conditions; B. Lineweaver-Burk plot of competition relationship between ML251 and ATP; C. Lineweaver-Burk plot of competition relationship between ML251 and F6P.
4.3. Planned Future Studies
ML251 was extensively profiled against relative targets. Our studies indicate that ML251 inhibits trypanosomatid isoforms of PFK and lacks cytotoxicity in a number of human cell lines. We could not test ML251 against purified human PFK because it is currently not commercially available. Should this change, this could provide valuable information and would be an experiment we would pursue.
Ongoing efforts remain to further elucidate binding modes of ML251 via co-crystallization with purified Tb PFK. Inevitably, this result will provide invaluable inspiration for the further development of an optimized inhibitor of this promising target. To help support on-target activity, well-validated strains of T. brucei that have been engineered to either over- or under-express PFK, will be used to examine how inhibitor potency is modulated by PFK levels.
Currently, there exists a divergence in SAR between the PFK enzymatic assay and standard cultured parasite assay. This is likely due to the inability of some compounds to access the target enzyme in the more complex, and highly compartmentalized, living trypanosome. As a result, the decision has been made to use the in vitro parasite assay as the driver for future analog and SAR development as a means to optimize potential for in vivo translation. Though this assay format is lower in throughput and more expensive than its enzymatic counterpart, efforts are underway to develop a protocol for use of the assay at either NCGC or the University of Edinburgh, which will aid in the development of an inhibitor possessing a desired ultimate potency (ED50 < 5 μM) and selectivity window (> 100-fold with respect to cytotoxicity in KB-3-1 cells). Should this benchmark be achieved, lead compounds will be advanced to in vivo mouse models of trypanosome infection. Such models have been well-characterized and are available in a fee-for-service format through multiple contract research organizations.
5. References
- 1.
- Nwaka S, Hudson A. Nat. Rev. Drug Discov. 2006;5:941–955. [PubMed: 17080030]
- 2.
- Verlinde CLMJ, Hannaert V, Blonski C, Willson M, Périé JJ, Fothergill-Gilmore LA, Opperdoes FR, Gelb MH, Hol WGJ, Michels PAM. Drug Res. Updates. 2001;4:50–65. [PubMed: 11512153]
- 3.
- Albert M.-A, Haanstra JR, Hannaert V, Van Roy J, Opperdoes FR, Bakker BM, Michels PA. J. Biol. Chem. 2005;280:28306–28315. [PubMed: 15955817]
- 4.
- Martinez-Oyanedel J, McNae IW, Nowicki MW, Keillor JW, Michels PA, Fothergill-Gilmore LA, Walkinshaw MD. J. Mol. Biol. 2007;366:1185–1198. [PubMed: 17207816]
- 5.
- Keillor JW, Lherbet C, Castonguay R, Lapierre D, Martinez-Oyanedel J, Fothergill-Gilmore LA, Walkinshaw MD. Acta Cryst. 2003;D59:532–534. [PubMed: 12595717]
- 6.
- Claustre S, Denier C, Lakhdar-Ghazal F, Lougare A, Lopez C, Chevalier N, Michels PA, Périé J, Willson M. Biochemistry. 2002;41:10183–10193. [PubMed: 12162732]
- 7.
- McNae IW, Martinez-Oyanedel J, Keillor JW, Michels PA, Fothergill-Gilmore LA, Walkinshaw MD. J. Mol. Biol. 2009;385:1519–1533. [PubMed: 19084537]
- 8.
- Willson M, Callens M, Kuntz DA, Périé J, Opperdoes FR. Mol. Biochem. Parasitol. 1993;59:201–210. [PubMed: 8341319]
- 9.
- Ngantchou I, Nyasse B, Denier C, Blonski C, Hannaert V, Schneider B. Bioorg. Med. Chem. Lett. 2010;20:3495–3498. [PubMed: 20529682]
- 10.
- Nowicki MW, Tulloch LB, Worrall L, McNae IW, Hannaert V, Michels PA, Fothergill-Gilmore LA, Walkinshaw MD, Turner NJ. Bioorg. Med. Chem. 2008;16:5050–5061. [PubMed: 18387804]
- 11.
- Rodriguez E, Lander N, Ramirez JL. Mem. Inst. Oswaldo Cruz. 2009;104:745–748. [PubMed: 19820836]
- 12.
- Croft SL, Snowdon D, Yardley V. J. Antimicrob. Chemother. 1996;38:1041–1047. [PubMed: 9023651]
- 13.
- Croft SL, Barrett MP, Urbina JA. Trends Parasitol. 2005;21:508–512. [PubMed: 16150644]
- 14.
- Lopez C, Chevalier N, Hannaert V, Rigden DJ, Michels PA, Ramirez JL. Eur. J. Biochem. 2002;269:3978–3989. [PubMed: 12180974]
- 15.
- Berg M, Van der Veken P, Joossens J, Muthusamy V, Breugelmans M, Moss CX, Rudolf J, Cos P, Coombs GH, Maes L, Haemers A, Mottram JC, Augustyns K. Bioorg Med Chem Lett. 2010:2001–2006. [PMC free article: PMC2845880] [PubMed: 20167486]
- 16.
- Spinks D, Ong HB, Mpamhanga CP, Shanks EJ, Robinson DA, Collie IT, Read KD, Frearson JA, Wyatt PG, Brenk R, Fairlamb AH, Gilbert IH. ChemMedChem. 2011;6:302–308. [PMC free article: PMC3047710] [PubMed: 21275054]
- 17.
- Otoguro K, Ishiyama A, Iwatsuki M, Namatame M, Nishihara-Tukashima Y, Omura S, Yamada H. J Nat Med. 2011. published online. [PubMed: 22116743]
Appendix
Probe Characterization
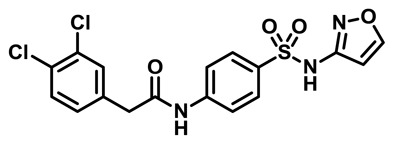
ML251: LC-MS Retention Time: t1 (Method 1) = 5.734 min and t2 (Method 2) = 3.624 min; 1H NMR (400 MHz, DMSO-d6) δ ppm 11.45 (1 H, br. s.), 10.60 (1 H, s), 8.70 (1 H, d, J=1.4 Hz), 7.78 – 7.83 (2 H, m), 7.72 – 7.78 (2 H, m), 7.55 – 7.62 (2 H, m), 7.30 (1 H, dd, J=8.3, 1.7 Hz), 6.40 (1 H, d, J=1.6 Hz), 3.73 (2 H, s); HRMS (ESI) m/z (M+H)+ calcd. for C17H14Cl2N3O4S, 426.0077; found 426.0077.
Analog Characterization

CID-53255380: LC-MS Retention Time: t1 (Method 1) = 5.395 min and t2 (Method 2) = 3.352 min; 1H NMR (400 MHz, DMSO-d6) d ppm 10.97 (1 H, br. s.), 7.55 (1 H, d, J=8.2 Hz), 7.49 (1 H, d, J=2.0 Hz), 7.20 (1 H, dd, J=8.2, 2.0 Hz), 6.12 (1 H, s), 4.37 – 4.52 (1 H, m), 4.00 – 4.13 (1 H, m), 3.69 – 3.83 (2 H, m), 3.46 – 3.59 (1 H, m), 3.02 – 3.14 (1 H, m), 2.57 – 2.69 (1 H, m), 2.34 (3 H, s), 1.90 – 2.04 (2 H, m), 1.35 – 1.61 (2 H, m); HRMS (ESI) m/z (M+H)+ calcd. for C17H20Cl2N3O4S, 432.0546; found 432.0549.

CID-5220784: LC-MS Retention Time: t1 (Method 1) = 5.159 min and t2 (Method 2) = 3.408 min; 1H NMR (400 MHz, DMSO-d6) d ppm 11.30 (1 H, s), 10.57 (1 H, s), 7.72 – 7.85 (4 H, m), 7.28 – 7.38 (4 H, m), 7.21 – 7.28 (1 H, m,), 6.10 (1 H, s), 3.67 (2 H, s), 2.28 (3 H, s); HRMS (ESI) m/z (M+H)+ calcd. for C18H18N3O4S, 372.1013; found 372.1020.

CID-2434603: LC-MS Retention Time: t1 (Method 1) = 4.855 min and t2 (Method 2) = 3.108 min; 1H NMR (400 MHz, DMSO-d6) d ppm 11.30 (1 H, br. s.), 10.50 (1 H, s), 7.73 – 7.81 (4 H, m), 6.93 (1 H, d, J=1.6 Hz), 6.86 – 6.91 (1 H, m), 6.80 – 6.85 (1 H, m), 6.10 (1 H, s), 3.73 (3 H, s), 3.72 (3 H, s), 3.58 (2 H, s), 2.28 (3 H, s); HRMS (ESI) m/z (M+H)+ calcd. for C20H22N3O6S, 432.1244; found 432.1229.

CID-53255430: LC-MS Retention Time: t1 (Method 1) = 5.636 min and t2 (Method 2) = 3.563 min; 1H NMR (400 MHz, DMSO-d6) d ppm 11.31 (1 H, br. s.), 10.59 (1 H, s), 7.72 – 7.82 (4 H, m), 7.53 (1 H, dd, J=8.2, 7.8 Hz), 7.37 (1 H, dd, J=10.4, 1.8 Hz), 7.18 (1 H, dd, J=8.2, 1.2 Hz), 6.10 (1 H, s), 3.74 (2 H, s), 2.28 (3 H, s); HRMS (ESI) m/z (M+H)+ calcd. for C18H16ClFN3O4S, 424.0529; found 424.0534.

CID-53255399: LC-MS Retention Time: t1 (Method 1) = 5.919 min and t2 (Method 2) = 3.693 min; 1H NMR (400 MHz, DMSO-d6) d ppm 10.60 (1 H, s), 7.73 – 7.85 (4 H, m), 7.53 – 7.65 (2 H, m), 7.31 (1 H, dd, J=8.4, 1.8 Hz), 5.62 (1 H, br. s.), 3.74 (2 H, s), 2.07 (3 H, s), amide proton not observed; HRMS (ESI) m/z (M+H)+ calcd. for C18H16Cl2N3O4S, 440.0233; found 440.0240.
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Identification of ML251, a Potent Inhibitor of T. brucei and T. cruzi Phosphofructokinase.[ACS Med Chem Lett. 2014]Identification of ML251, a Potent Inhibitor of T. brucei and T. cruzi Phosphofructokinase.Brimacombe KR, Walsh MJ, Liu L, Vásquez-Valdivieso MG, Morgan HP, McNae I, Fothergill-Gilmore LA, Michels PA, Auld DS, Simeonov A, et al. ACS Med Chem Lett. 2014 Jan 9; 5(1):12-7. Epub 2013 Oct 30.
- Review Identification of Inhibitors of Trypanosoma brucei Hexokinases.[Probe Reports from the NIH Mol...]Review Identification of Inhibitors of Trypanosoma brucei Hexokinases.Sharlow E, Golden JE, Dodson H, Morris M, Hesser M, Lyda T, Leimgruber S, Schroeder CE, Flaherty DP, Weiner WS, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Review [Development of antituberculous drugs: current status and future prospects].[Kekkaku. 2006]Review [Development of antituberculous drugs: current status and future prospects].Tomioka H, Namba K. Kekkaku. 2006 Dec; 81(12):753-74.
- Application of a resazurin-based high-throughput screening assay for the identification and progression of new treatments for human African trypanosomiasis.[Int J Parasitol Drugs Drug Res...]Application of a resazurin-based high-throughput screening assay for the identification and progression of new treatments for human African trypanosomiasis.Bowling T, Mercer L, Don R, Jacobs R, Nare B. Int J Parasitol Drugs Drug Resist. 2012 Dec; 2:262-70. Epub 2012 Mar 3.
- Drug discovery for human African trypanosomiasis: identification of novel scaffolds by the newly developed HTS SYBR Green assay for Trypanosoma brucei.[J Biomol Screen. 2015]Drug discovery for human African trypanosomiasis: identification of novel scaffolds by the newly developed HTS SYBR Green assay for Trypanosoma brucei.Faria J, Moraes CB, Song R, Pascoalino BS, Lee N, Siqueira-Neto JL, Cruz DJ, Parkinson T, Ioset JR, Cordeiro-da-Silva A, et al. J Biomol Screen. 2015 Jan; 20(1):70-81. Epub 2014 Oct 23.
- Identification of Selective Inhibitors of Phosphofructokinase as Lead Compounds ...Identification of Selective Inhibitors of Phosphofructokinase as Lead Compounds Against Trypanosomiasis - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...