Summary
Clinical characteristics.
Autosomal dominant craniometaphyseal dysplasia (designated AD-CMD in this review) is characterized by progressive diffuse hyperostosis of cranial bones evident clinically as wide nasal bridge, paranasal bossing, widely spaced eyes with an increase in bizygomatic width, and prominent mandible. Development of dentition may be delayed and teeth may fail to erupt as a result of hyperostosis and sclerosis of alveolar bone. Progressive thickening of craniofacial bones continues throughout life, often resulting in narrowing of the cranial foramina, including the foramen magnum. If untreated, compression of cranial nerves can lead to disabling conditions such as facial palsy, blindness, or deafness (conductive and/or sensorineural hearing loss). In individuals with typical uncomplicated AD-CMD life expectancy is normal; in those with severe AD-CMD life expectancy can be reduced as a result of compression of the foramen magnum.
Diagnosis/testing.
Diagnosis is based on clinical and radiographic findings that include diffuse hyperostosis of the cranial base, cranial vault, facial bones, and mandible as well as widening and radiolucency of metaphyses in long bones. Identification of a heterozygous pathogenic variant in ANKH by molecular genetic testing can confirm the diagnosis if clinical features are inconclusive.
Management.
Treatment of manifestations: Treatment for feeding and respiratory issues per craniofacial team; surgical intervention to reduce compression of cranial nerves and the brain stem / spinal cord at the level of the foramen magnum. Severely overgrown facial bones can be contoured; however, surgical procedures can be technically difficult and bone regrowth is common. Hearing aids; vision aids and surgical treatment for optic nerve impaction; speech therapy; surgical intervention for malocclusion.
Surveillance: Evaluation for feeding and respiratory issues at least annually. Neurologic evaluation for signs and symptoms of narrowing of the cranial foramina including the foramen magnum at least annually. Hearing and ophthalmologic assessment at least annually.
Genetic counseling.
By definition, AD-CMD is inherited in an autosomal dominant manner. Most individuals diagnosed with AD-CMD have an affected parent; the proportion of individuals with AD-CMD caused by a de novo pathogenic variant is thought to be very low. Each child of an individual with AD-CMD has a 50% chance of inheriting the pathogenic variant. Once the AD-CMD-causing pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
Formal diagnostic criteria for autosomal dominant craniometaphyseal dysplasia (AD-CMD) have not been established.
Suggestive Findings
AD-CMD should be suspected in individuals with the following clinical, radiographic, and laboratory features.
Clinical features
- Obstruction of the nasal sinuses
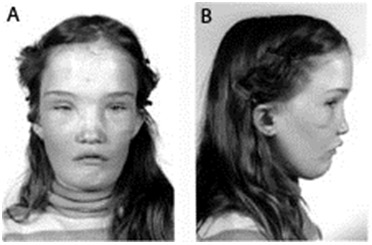
- Characteristic facial features. Wide nasal bridge, paranasal bossing, hypertelorism with an increase in bizygomatic width, and prominent mandible (see Figure 1)
- Dolichocephaly due to fronto-occipital hyperostosis

Figure 1.
Facial features of a girl age 13 years with AD-CMD Reprinted from Reichenberger et al [2001] with permission from Elsevier
Radiographic features
- Cranial base. Sclerosis may begin in infancy (see Figure 2). Increasing diffuse hyperostosis of the cranial base leads to narrowing of the foramen magnum.
- Skull. Diffuse hyperostosis of cranial vault, facial bones, and mandible increases as the condition progresses [Lamazza et al 2009] with obstruction of the cranial foramina.
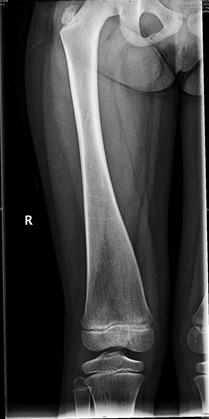
- Long bones. Metaphyseal widening (described as Erlenmeyer flask- or club-shaped) with thinned cortex and decreased bony density in the metaphyses can be detected early in life. Metaphyseal changes typically develop during early childhood. The flaring is most prominent in the distal femur and tibia (see Figure 3). Diaphyseal sclerosis/hyperostosis can be present in infancy but disappears with age. Bone density of the diaphyses is normal in children and adults; cortical thickness can be increased.
- Ribs and clavicles (medial portion [i.e., endochondral]) can be sclerotic in younger children but show normal bone density by age five years [Richards et al 1996].

Figure 2.
Increased thickness of craniofacial bones in a child age three years with AD-CMD

Figure 3.
Metaphyseal widening of long bones, specifically prominent at the knee joint
Laboratory features
- Blood calcium and phosphate concentrations are within normal limits [Cheung et al 1997] or decreased [Sheppard et al 2003].
- Serum alkaline phosphatase activity can be elevated [Sheppard et al 2003, Wu et al 2016].
- Parathyroid hormone level is normal or can be slightly/transiently elevated [Fanconi et al 1988, Cheung et al 1997, Sheppard et al 2003, Wu et al 2016].
- Osteocalcin is decreased [Yamamoto et al 1993].
Note: Findings are based on very limited data. Variability of the described parameters can be expected. Abnormal parameters may be transient.
Establishing the Diagnosis
The diagnosis of AD-CMD is established in a proband with characteristic craniofacial hyperostosis and flaring and undertrabeculation of long bone metaphyses and/or a heterozygous pathogenic variant in ANKH identified by molecular genetic testing (see Table 1).
Note: Identification of a heterozygous ANKH variant of uncertain significance does not establish or rule out the diagnosis of this disorder.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing and multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of AD-CMD is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with hyperostosis are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of AD-CMD, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of ANKH is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications. Note: To date such variants have not been identified as a cause of this disorder.
- A multigene panel that includes ANKH and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by hyperostosis, comprehensive genomic testing, which does not require the clinician to determine which gene is likely involved, is most likely to establish the diagnosis. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic – and particularly when evidence supports autosomal dominant inheritance – exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis. Note: To date such variants have not been identified as a cause of this disorder.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Autosomal Dominant Craniometaphyseal Dysplasia
Clinical Characteristics
Clinical Description
Autosomal dominant craniometaphyseal dysplasia (AD-CMD) is often detected within the first few weeks of life because of breathing or feeding problems resulting from choanal stenosis (narrowing of nasal sinus) [Haverkamp et al 1996, Cheung et al 1997, Taggart et al 2014].
Early stages of AD-CMD can be radiographically recognized as sclerosis of the cranial base. Hyperostosis of the cranial base, cranial vault, facial bones, and mandible occurs gradually. Overgrowth of the lower jaw (mandibular hyperostosis) and recessed midface (midface retrusion) are often seen [Hayashibara et al 2000].
Progressive thickening of craniofacial bones continues throughout life, often resulting in narrowing of the cranial foramina, including the foramen magnum. If untreated, compression of cranial nerves can lead to disabling conditions such as facial palsy, blindness, or deafness (conductive and/or sensorineural hearing loss) as cranial hyperostosis and sclerosis progress [Beighton et al 1979, Richards et al 1996]. Nasal obstruction and mandibular hyperostosis affect speech modulation.
Associated Chiari I malformation can lead to severe headaches [Tanaka et al 2013].
Development of dentition may be delayed and teeth may fail to erupt as a result of hyperostosis and sclerosis of alveolar bone [Chen et al 2014].
Malocclusion and anterior cross-bite can be caused by jaw overgrowth [Hayashibara et al 2000].
Life expectancy. Individuals with typical uncomplicated AD-CMD have normal life expectancy. Expressivity in simplex cases (i.e., single occurrence in a family) of CMD is highly variable.
Genotype-Phenotype Correlations
No genotype-phenotype correlation has been reported.
The phenotypic severity (expressivity) in AD-CMD is variable even among affected members of the same family.
Penetrance
Penetrance is 100%. Males and females are equally affected.
Nomenclature
AD-CMD was previously referred to as "craniometaphyseal dysplasia-Jackson type."
Prevalence
CMD is very rare. No epidemiology has been established.
Genetically Related (Allelic) Disorders
Table 2.
ANKH Allelic Disorders
Differential Diagnosis
Table 3.
Genes of Interest in the Differential Diagnosis of Autosomal Dominant Craniometaphyseal Dysplasia
Braun-Tinschert type of metaphyseal dysplasia (OMIM 605946) is inherited in an autosomal dominant manner. The gene(s) in which mutation is causative are unknown [Braun et al 2001].
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with autosomal dominant craniometaphyseal dysplasia (AD-CMD), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Autosomal Dominant Craniometaphyseal Dysplasia
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with Autosomal Dominant Craniometaphyseal Dysplasia
Surveillance
Table 6.
Recommended Surveillance for Individuals with Autosomal Dominant Craniometaphyseal Dysplasia
Evaluation of Relatives at Risk
It is appropriate to evaluate relatives at risk in order to identify as early as possible those who would benefit from initiation of treatment and preventive measures. Early diagnosis of at-risk relatives may be beneficial for management of complications from progressive hyperostosis.
Evaluations can include:
- Molecular genetic testing if the pathogenic variant in the family is known;
- Clinical evaluation and cranial and long bone radiographs if the pathogenic variant in the family is not known.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Other
Calcitonin has been thought to be effective because of its inhibitory effect on bone turnover. However, previous case reports found calcitonin therapy to be ineffective in treating hyperplasia of craniofacial bones in persons with CMD [Fanconi et al 1988, Haverkamp et al 1996].
Calcitriol with a low-calcium diet to stimulate bone resorption by promoting osteoclast formation had been reported to improve facial paralysis but has no effect on metaphyseal deformity [Key et al 1988, Wu et al 2016].
Acetazolamide has been suggested for treatment of disorders with increased bone mineral density. González-Rodríguez et al [2016] reported acetazolamide use in an individual with a phenotype similar to CMD, but diagnosis of AD-CMD was not confirmed.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
By definition, autosomal dominant craniometaphyseal dysplasia (AD-CMD) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Most individuals diagnosed with AD-CMD have an affected parent.
- Some individuals diagnosed with AD-CMD have the disorder as the result of a de novo pathogenic variant. The proportion of individuals with AD-CMD caused by a de novo pathogenic variant is thought to be very low; however statistical data are not available.
- If the proband appears to be the only affected family member (i.e., a simplex case) and has a known ANKH pathogenic variant, molecular genetic testing is recommended for the parents of a proband.
- If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent.* Though theoretically possible, no instances of a proband inheriting a pathogenic variant from a parent with germline mosaicism have been reported.* Misattributed parentage can also be explored as an alternative explanation for an apparent de novo pathogenic variant.
- The family history of some individuals diagnosed with AD-CMD may appear to be negative because of failure to recognize the disorder in affected family members. Therefore, an apparently negative family history cannot be confirmed without appropriate clinical evaluation of the parents and/or molecular genetic testing (to establish that neither parent is heterozygous for the pathogenic variant identified in the proband).
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs is 50%. Because penetrance of AD-CMD is 100%, sibs who inherit a pathogenic variant will develop the phenotype although the severity of the phenotype may vary.
- If the proband has a known ANKH pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If the genetic status of the parents is unknown but they are clinically unaffected, the risk to the sibs of a proband appears to be low but still increased over that of the general population because of the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with AD-CMD has a 50% chance of inheriting the causative pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: If a parent is affected, his or her family members may be at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
Prenatal Testing and Preimplantation Genetic Testing
Once the AD-CMD-causing pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- American Society for Deaf ChildrenPhone: 800-942-2732 (ASDC)Email: info@deafchildren.org
- Children's Craniofacial AssociationPhone: 800-535-3643Email: contactCCA@ccakids.com
- Face Equality InternationalUnited Kingdom
- National Association of the DeafPhone: 301-587-1788 (Purple/ZVRS); 301-328-1443 (Sorenson); 301-338-6380 (Convo)Fax: 301-587-1791Email: nad.info@nad.org
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Craniometaphyseal Dysplasia, Autosomal Dominant: Genes and Databases
Table B.
OMIM Entries for Craniometaphyseal Dysplasia, Autosomal Dominant (View All in OMIM)
Molecular Pathogenesis
ANKH encodes the progressive ankylosis protein homolog, a multispan transmembrane protein located at the outer cell membrane that transports intracellular pyrophosphate into the extracellular matrix. Pyrophosphate is a regulator of matrix (bone) mineralization. The protein sequence of the progressive ankylosis protein homolog is highly conserved among vertebrate animals.
Progressive ankylosis protein homolog expressing a craniometaphyseal dysplasia-associated variant most likely has a reduced ability to transport intracellular pyrophosphate from osteoblasts to the bone matrix [Ho et al 2000].
Mechanism of disease causation. Most common pathogenic variants result in one-amino acid deletions, while others are missense or small in-frame deletions and insertions [Nürnberg et al 2001, Reichenberger et al 2001, Kornak et al 2010, Zajac et al 2010, Dutra et al 2012]. Most pathogenic variants occur in the nucleotide region encoding presumed intracellular domains of the transmembrane loop structure. Based on findings in knockout and knock-in mice studies, ANKH pathogenic variants are thought to result in a dominant negative gain of function as well as loss of function of pyrophosphate transport. The shared phenotype between these murine models is explained by the rapid degradation of pathogenic ANK protein [Kanaujiya et al 2018]. To date, no other information on mechanism is available.
References
Literature Cited
- Baynam G, Goldblatt J, Schofield L. Craniometaphyseal dysplasia and chondrocalcinosis cosegregating in a family with an ANKH mutation. Am J Med Genet A. 2009;149A:1331–3. [PubMed: 19449425]
- Beighton P, Hamersma H, Horan F. Craniometaphyseal dysplasia--variability of expression within a large family. Clin Genet. 1979;15:252–8. [PubMed: 421364]
- Braun HS, Nurnberg P, Tinschert S. Metaphyseal dysplasia: a new autosomal dominant type in a large German kindred. Am J Med Genet. 2001;101:74–7. [PubMed: 11343343]
- Chen IP, Tadinada A, Dutra EH, Utrja A, Uribe F, Reichenberger EJ. Dental anomalies associated with craniometaphyseal dysplasia. J Dent Res. 2014;93:553–8. [PMC free article: PMC4023465] [PubMed: 24663682]
- Cheung VG, Boechat MI, Barrett CT. Bilateral choanal narrowing as a presentation of craniometaphyseal dysplasia. J Perinatol. 1997;17:241–3. [PubMed: 9210083]
- Dutra EH, Chen I-P, McGregor TL, Ranells JD, Reichenberger EJ. Two novel large ANKH deletion mutations in sporadic cases with craniometaphyseal dysplasia. Clin Genet. 2012;81:93–5. [PMC free article: PMC3417334] [PubMed: 22150416]
- Fanconi S, Fischer JA, Wieland P, Giedion A, Boltshauser E, Olah AJ, Landolt AM, Prader A. Craniometaphyseal dysplasia with increased bone turnover and secondary hyperparathyroidism: therapeutic effect of calcitonin. J Pediatr. 1988;112:587–91. [PubMed: 3351685]
- González-Rodríguez JD, Luis-Yanes MI, Ingles-Torres E, Arango-Sancho P, Cabrera-Sevilla JE, Duque-Fernandez MR, Gil-Sanchez S, Garcia-Nieto VM. Can acetazolamide be used to treat diseases involving increased bone mineral density? Intractable Rare Dis Res. 2016;5:284–9. [PMC free article: PMC5116865] [PubMed: 27904825]
- Haverkamp F, Emons D, Straehler-Pohl HJ, Zerres K. Craniometaphyseal dysplasia as a rare cause of a severe neonatal nasal obstruction. Int J Pediatr Otorhinolaryngol. 1996;34:159–64. [PubMed: 8770684]
- Hayashibara T, Komura T, Sobue S, Ooshima T. Tooth eruption in a patient with craniometaphyseal dysplasia: case report. J Oral Pathol Med. 2000;29:460–2. [PubMed: 11016689]
- Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–70. [PubMed: 10894769]
- Kanaujiya J, Bastow E, Luxmi R, Hao Z, Zattas D, Hochstrasser M, Reichenberger EJ, Chen IP. Rapid degradation of progressive ankylosis protein (ANKH) in craniometaphyseal dysplasia. Scientific Reports. 2018;8:15710. [PMC free article: PMC6200807] [PubMed: 30356088]
- Key LL Jr, Volberg F, Baron R, Anast CS. Treatment of craniometaphyseal dysplasia with calcitriol. J Pediatr. 1988;112:583–7. [PubMed: 3351684]
- Kornak U, Brancati F, Le Merrer M, Lichtenbelt K, Hohne W, Tinschert S, Garaci FG, Dallapiccola B, Nurnberg P. Three novel mutations in the ANK membrane protein cause craniometaphyseal dysplasia with variable conductive hearing loss. Am J Med Genet A. 2010;152A:870–4. [PubMed: 20358596]
- Lamazza L, Messina A, D'Ambrosio F, Spink M, De Biase A. Craniometaphyseal dysplasia: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107:e23–7. [PubMed: 19426903]
- Morava E, Kühnisch J, Drijvers JM, Robben JH, Cremers C, van Setten P, Branten A, Stumpp S, de Jong A, Voesenek K, Vermeer S, Heister A, Claahsen-van der Grinten HL, O'Neill CW, Willemsen MA, Lefeber D, Deen PM, Kornak U, Kremer H, Wevers RA. Autosomal recessive mental retardation, deafness, ankylosis, and mild hypophosphatemia associated with a novel ANKH mutation in a consanguineous family. J Clin Endocrinol Metab. 2011;96:E189–98. [PMC free article: PMC5393418] [PubMed: 20943778]
- Nürnberg P, Thiele H, Chandler D, Höhne W, Cunningham ML, Ritter H, Leschik G, Uhlmann K, Mischung C, Harrop K, Goldblatt J, Borochowitz ZU, Kotzot D, Westermann F, Mundlos S, Braun HS, Laing N, Tinschert S. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet. 2001;28:37–41. [PubMed: 11326272]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C, Baur ST, Shiang R, Grange DK, Beighton P, Gardner J, Hamersma H, Sellars S, Ramesar R, Lidral AC, Sommer A, Raposo do Amaral CM, Gorlin RJ, Mulliken JB, Olsen BR. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet. 2001;68:1321–6. [PMC free article: PMC1226118] [PubMed: 11326338]
- Richards A, Brain C, Dillon MJ, Bailey CM. Craniometaphyseal and craniodiaphyseal dysplasia, head and neck manifestations and management. J Laryngol Otol. 1996;110:328–38. [PubMed: 8733453]
- Sheppard WM, Shprintzen RJ, Tatum SA, Woods CI. Craniometaphyseal dysplasia: a case report and review of medical and surgical management. Int J Pediatr Otorhinolaryngol. 2003;67:71–7. [PubMed: 12560153]
- Taggart MG, Crockett DJ, Meier JD, Wiggins RH. Infant with persistent nasal obstruction. JAMA Otolaryngol Head Neck Surg. 2014;140:983–4. [PubMed: 25188335]
- Tanaka M, Arataki S, Sugimoto Y, Takigawa T, Tetsunaga T, Ozaki T. Chiari type I malformation caused by craniometaphyseal dysplasia. Acta Med Okayama. 2013;67:385–9. [PubMed: 24356723]
- Wu B, Jiang Y, Wang O, Li M, Xing XP, Xia WB. Craniometaphyseal dysplasia with obvious biochemical abnormality and rickets-like features. Clin Chim Acta. 2016;456:122–7. [PubMed: 26820766]
- Yamamoto T, Kurihara N, Yamaoka K, Ozono K, Okada M, Yamamoto K, Matsumoto S, Michigami T, Ono J, Okada S. Bone marrow-derived osteoclast-like cells from a patient with craniometaphyseal dysplasia lack expression of osteoclast-reactive vacuolar proton pump. J Clin Invest. 1993;91:362–7. [PMC free article: PMC330035] [PubMed: 7678608]
- Zajac A, Baek SH, Salhab I, Radecki MA, Kim S, Hakonarson H, Nah HD. Novel ANKH mutation in a patient with sporadic craniometaphyseal dysplasia. Am J Med Genet A. 2010;152A:770–6. [PMC free article: PMC2944898] [PubMed: 20186813]
Chapter Notes
Revision History
- 11 June 2020 (sw) Comprehensive update posted live
- 15 January 2015 (me) Comprehensive update posted live
- 2 November 2010 (me) Comprehensive update posted live
- 27 August 2007 (me) Review posted live
- 25 May 2007 (er) Original submission
Publication Details
Author Information and Affiliations
Center for Regenerative Medicine and Skeletal Development
UConn Health (UCH)
Farmington, Connecticut
UConn Health (UCH)
Farmington, Connecticut
Publication History
Initial Posting: August 27, 2007; Last Update: June 11, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Reichenberger E, Chen IP. Craniometaphyseal Dysplasia, Autosomal Dominant. 2007 Aug 27 [Updated 2020 Jun 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.