Summary
Clinical characteristics.
Au-Kline syndrome is characterized by developmental delay and hypotonia with moderate-to-severe intellectual disability, and typical facial features that include long palpebral fissures, ptosis, shallow orbits, large and deeply grooved tongue, broad nose with a wide nasal bridge, and downturned mouth. Congenital heart disease, hydronephrosis, palate abnormalities, and oligodontia are reported in the majority of affected individuals. Variable autonomic dysfunction (gastrointestinal dysmotility, high pain threshold, heat intolerance, recurrent fevers, abnormal sweating) is found in more than one third of affected individuals. Additional complications can include craniosynostosis, feeding difficulty, vision issues, hearing loss, osteopenia, and other skeletal anomalies. Epilepsy and brain malformations are rare.
Diagnosis/testing.
The diagnosis of Au-Kline syndrome is established in a proband by identification of a heterozygous pathogenic variant in HNRNPK on molecular genetic testing.
Management.
Treatment of manifestations: Physical therapy may be helpful for hypotonia. Feeding therapy for poor weight gain; gastrostomy tube placement may be required for persistent feeding issues. Referral to neurologist with experience in management of autonomic dysfunction. Bisphosphonate treatment could be considered for those with osteopenia who experience recurrent fractures. Standard treatment for developmental delay / intellectual disability, behavior concerns, epilepsy, craniosynostosis, palatal anomalies, congenital heart defects / aortic dilatation / cardiomyopathy, hydronephrosis, cryptorchidism, bowel dysfunction, skeletal anomalies, refractive errors, keratopathy, hearing loss, malocclusion / open bite, oligodontia, hypothyroidism, hypoventilation, and obstructive sleep apnea.
Prevention of secondary complications: Anesthesia consultation is suggested prior to any sedation for surgery given potential airway issues from malocclusion and macroglossia. There is also potential risk that prolonged intubation and ventilation will be required, as occurred in one individual after surgery.
Surveillance: At each visit, measure growth parameters and evaluate nutritional status; monitor developmental progress and educational needs; assess for neurobehavioral/psychiatric manifestations; monitor those with seizures; assess for signs and symptoms of sleep apnea. At all health visits in the first few months of life, screen for craniosynostosis. At all health visits and at least annually, assess for new manifestations, such as seizures or dysautonomia; clinical examination for scoliosis. At least every 6 months, dental and/or orthodontic evaluation. Annually, TSH and free T4. Annually or as clinically indicated, audiology evaluation. The frequency of echocardiogram and assessment for cardiomyopathy should be determined by a cardiologist. The frequency of ophthalmology evaluations should be determined by an ophthalmologist. Consideration of bone densitometry is based on the severity of osteopenia and history of fractures.
Genetic counseling.
Au-Kline syndrome is inherited in an autosomal dominant manner. All probands reported to date with Au-Kline syndrome have the disorder as a result of a de novo HNRNPK pathogenic variant. Each child of an individual with Au-Kline syndrome has a 50% chance of inheriting the HNRNPK pathogenic variant. Prenatal testing for a pregnancy at increased risk is possible if the HNRNPK pathogenic variant in the family is known.
Diagnosis
Clinical diagnostic criteria for Au-Kline syndrome (AKS) have been proposed [Choufani et al 2022].
Suggestive Findings
AKS should be suspected in individuals with at least four of the following six characteristic facial features (see Figure 1):

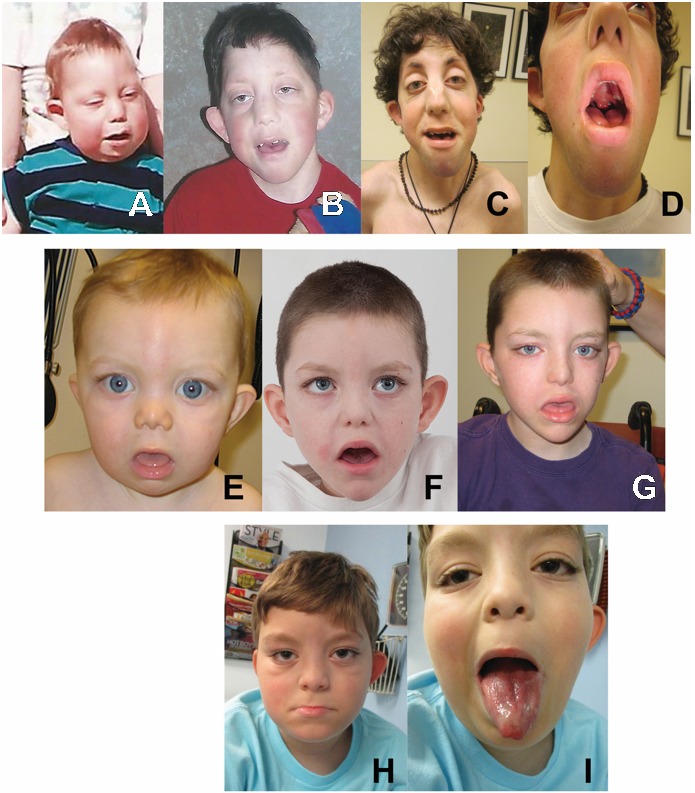
Figure 1.
Three individuals with Au-Kline syndrome A-C. Proband 1 at age 14 months, age eight years, and age 19 years. Note long palpebral fissures, prominent eyes and shallow orbits, broad nasal ridge, and M-shaped Cupid's bow to upper lip.
- Long palpebral fissures
- Ptosis
- Shallow orbits
- Deeply grooved tongue
- Broad nose with wide nasal bridge and thick alae nasi
- Downturned mouth, often described as an M-shaped Cupid's bow
AND
- Global developmental delay or intellectual disability (defined as a DQ or IQ score below 70)
- Hypotonia
A diagnosis is considered highly likely in an individual with at least five of the six characteristic facial features in addition to global developmental delay / intellectual disability and hypotonia [Au et al 2015, Au et al 2018, Choufani et al 2022]; however, molecular genetic testing (see Establishing the Diagnosis) is still recommended.
Further supportive features may include:
- Craniosynostosis (Typically, sagittal and metopic sutures are affected; metopic ridging is common.)
- Palate abnormalities (e.g., cleft palate, high-arched palate, bifid uvula)
- Congenital heart malformations (e.g., ventricular septal defect, atrial septal defect, bicuspid aortic valve, and more complex malformations)
- Genitourinary anomalies (e.g., hydronephrosis, undescended testes)
- Skeletal anomalies (e.g., vertebral segmentation defects, scoliosis, congenital hip dysplasia)
Establishing the Diagnosis
The diagnosis of AKS is established in a proband by identification of a heterozygous pathogenic (or likely pathogenic) variant in HNRNPK on molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) The identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis, although a distinctive epigenetic signature of Au-Kline syndrome has been identified (see Epigenetic signature analysis / methylation array).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing, exome array) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of Au-Kline syndrome is often recognizable, individuals with the distinctive findings described in Suggestive Findings may be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of Au-Kline syndrome has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of Au-Kline syndrome, molecular genetic testing approaches can include single-gene testing or use of a multigene panel.
- Single-gene testing. Sequence analysis of HNRNPK detects missense, nonsense, and splice site variants and small intragenic deletions/insertions; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis first. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.Note: Given the rarity of Au-Kline disorder, single-gene testing for HNRNPK may not be clinically available. A custom multigene panel or comprehensive genomic testing, or reanalysis of previous genomic testing data, may be required for HNRNPK analysis.
- A multigene panel that includes HNRNPK and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Of note, given the rarity of Au-Kline syndrome, some panels for intellectual disability may not include this gene. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1).
Option 2
When the diagnosis of Au-Kline syndrome is not considered because an individual has atypical phenotypic features or mild features that do not meet suggestive clinical diagnostic criteria, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Epigenetic signature analysis / methylation array. A distinctive epigenetic signature (disorder-specific genome-wide changes in DNA methylation profiles) in peripheral blood leukocytes has been identified in individuals with Au-Kline syndrome [Choufani et al 2022]. Epigenetic signature analysis of a peripheral blood sample or DNA banked from a blood sample can therefore be considered to clarify the diagnosis in individuals with: (1) suggestive findings of Au-Kline syndrome but in whom no pathogenic HNRNPK variant has been identified via sequence analysis or genomic testing; or (2) suggestive findings of Au-Kline syndrome and an HNRNPK variant of unknown clinical significance identified by molecular genetic testing. For an introduction to epigenetic signature analysis, click here.
Table 1.
Molecular Genetic Testing Used in Au-Kline Syndrome
Clinical Characteristics
Clinical Description
To date, at least 75 individuals have been identified with a pathogenic variant in HNRNPK [Au et al 2015, Lange et al 2016, Au et al 2018, Dentici et al 2018, Choufani et al 2022]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Select Features of Au-Kline Syndrome
Developmental delay (DD) and intellectual disability (ID). All affected individuals to date have been found to have developmental delay and/or intellectual disability. However, the degree of delay can be variable. Individuals with loss-of-function HNRNPK pathogenic variants typically have moderate-to-severe intellectual disability, whereas developmental outcome in those with missense pathogenic variants may be better [Au et al 2018; Choufani et al 2022; Authors, personal communication]. Autism spectrum disorder appears to be rare.
- In a group of older individuals with loss-of-function pathogenic variants (ranging in age from eight years to young adulthood), independent ambulation was achieved by 5/11 individuals, although some still required assistive devices for longer distances.Nine out of 12 individuals used verbal communication, with 6/12 using mostly single words and 3/12 using phrases to communicate.Eight out of 11 individuals were able to use sign language or communication devices to supplement their communication.
- In nine older individuals with missense pathogenic variants, 8/9 were able to walk independently, with 6/9 achieving this skill before age three years. Six out of eight individuals in this group were able to speak in phrases.
- Many older children and adults are also able to use signs (sometimes up to several hundred) and devices to supplement their communication.
Neurobehavioral/psychiatric manifestations. In individuals where information was available, autism spectrum disorder was reported in 2/13, attention-deficit/hyperactivity disorder in 2/14, anxiety in 2/14, obsessive-compulsive disorder in 1/14, and issues with impulse control in 1/14.
Other neurologic features
- Hypotonia has been reported in all known individuals with Au-Kline syndrome (AKS) and often persists into adulthood.
- Reflexes are typically reduced or absent.
- Some individuals have suspected neuropathy on exam, but formal nerve conduction studies / electromyography have not typically been pursued.
- Some individuals describe muscle weakness and/or easy fatigability.
- Muscle biopsy may be abnormal but has not been pursued in most individuals.
- Histologic findings have not been consistent and have included type 1 fiber atrophy and mitochondrial complex 1 deficiency.
- Several individuals have been diagnosed with autonomic dysfunction, presenting with gastrointestinal dysmotility, high pain threshold (37%), heat intolerance, recurrent fevers, and abnormal sweating.
Epilepsy. Seizures are rare, present in only 3% of affected individuals. For three individuals in which seizure information is available, absence seizures were reported in one and partial complex epilepsy in another. Seizures were controlled with anti-seizure medication in both individuals. The third individual had seizures that resolved.
Neuroimaging. Brain anomalies have been identified in several individuals. The most common abnormalities are gray matter heterotopia and thinning (hypoplasia) of the corpus callosum [Lange et al 2016, Au et al 2018]. Other findings can include spinal syrinx (particularly in those who also have scoliosis) and hypomyelination.
Craniofacial
- Craniosynostosis is present in approximately one quarter of individuals with AKS who have loss-of-function pathogenic variants but is rare in those who have pathogenic missense variants.Sagittal and metopic sutures are typically affected, and many individuals have metopic ridging without obvious or confirmed synostosis.Approximately half of the individuals with craniosynostosis have required surgical intervention. However, it is unclear if surgical intervention improves cognitive outcome in individuals with AKS.
- Palate abnormalities are common. Cleft palate has only been reported in individuals with loss-of-function pathogenic variants to date. Other palate anomalies, including bifid uvula and high-arched or narrow palate, have been observed in both genotypic groups (those with loss-of-function pathogenic variants and those with missense pathogenic variants; see Genotype-Phenotype Correlations).
- Typical facial features (see Figure 1). In the majority of individuals, the face is long, the orbits are shallow, and the palpebral fissures are long. There can be lateral lid eversion of the eyelids similar to Kabuki syndrome, but typically the lid eversion in AKS is more subtle. Ptosis is common and can be asymmetric. Ears can be protruding, with a simplified helix. Preauricular pits are common. The nose often has a characteristic shape with broad nasal bridge and tip, and occasionally thick alae nasi with notched nares. The mouth is frequently downturned and held in an open position. The upper lip is often described as an M-shaped Cupid's bow [Dentici et al 2018]. Many individuals have a deep midline groove in the tongue, and/or a bifid tip to the tongue. Many individuals also have macroglossia. Facial features may appear coarse. The face may appear rounder in infancy and elongates with time. Some individuals may have subtle facial findings that are not easily recognizable.
Cardiovascular. Congenital heart disease is present in approximately 63% of individuals with AKS (see Genotype-Phenotype Correlations).
- Ventricular septal defects are the most common anomaly.
- Complex congenital heart defects are rarely reported.
- Aortic dilatation has been identified in three individuals; the natural history of aortic dilatation in AKS is unclear.
- Two affected individuals have had left ventricular non-compaction cardiomyopathy.
Genitourinary
- Hydronephrosis is present in up to 52% of individuals with AKS (see Genotype-Phenotype Correlations). It is often identified prenatally and typically associated with vesicoureteral reflux or obstructive uropathy. It can sometimes be severe, as in one individual who was affected with prune-belly syndrome [Au et al 2018].
- Cryptorchidism is common in males.
Gastrointestinal complications and feeding. Most newborns are able to feed normally. Some individuals struggle with feeding difficulty and may require short- or long-term support with tube feeding (i.e., two individuals have needed long-term G-tube support). These issues may be associated with bowel dysmotility (e.g., delayed gastric emptying, recurrent vomiting, pseudo-obstruction). Constipation is common and can range from mild to severe.
Growth
- Most neonates with AKS have normal growth parameters.
- Approximately 30% of affected individuals demonstrate growth deficiency over time, affecting both height and weight.
- Age of onset of growth restriction has been variable, and the true incidence and degree of severity is unclear, as stature is also often affected by the presence of scoliosis.
- Occasionally, there may be mild microcephaly, which is usually noted by early childhood (see Genotype-Phenotype Correlations).
- Overgrowth consisting of increased length and weight at birth has been described in two individuals; two other individuals have been reported to have obesity with increased weight versus height [Au et al 2018].
Skeletal
- Thirty-nine percent of individuals with AKS have scoliosis, which can range from mild to severe and can require surgical intervention.
- Vertebral segmentation anomalies may be present and are more likely to be associated with severe scoliosis.
- Congenital hip dysplasia is observed in about one third of individuals with AKS.
- Talipes equinovarus and pes planus are also common.
- Some individuals have joint hypermobility, although joint dislocations have not been reported.
- Contractures are also occasionally reported, typically affecting large joints.
- Postaxial polydactyly has also been rarely reported.
Ears/hearing
- Hearing loss is present in approximately one quarter of affected individuals.Conductive hearing loss may be due to chronic middle ear effusion, but sensorineural hearing loss has been described in three individuals.
- Branchial defects have also been rarely reported.
Ophthalmologic
- Myopia and hyperopia have both been reported in individuals with AKS.
- Optic nerve anomalies, including hypoplasia of the optic nerve or the presence of a coloboma, have been identified in four affected individuals.
- There is theoretical risk for exposure keratopathy in individuals with particularly shallow orbits and long palpebral fissures, but this has not been observed in individuals with AKS.
Dental. The majority of affected individuals have malocclusion, and some have an open bite. Oligodontia is common. Bruxism is frequently observed.
Other associated features
- Endocrine. Osteopenia has been identified in several affected individuals; fractures have been seen in two individuals with AKS. Hypothyroidism is also present in several individuals.
- Respiratory. Most individuals with AKS who have been assessed have had normal sleep studies. One individual was reported to hypoventilate at night and required bilevel positive airway pressure. One individual was reported to have obstructive sleep apnea.
- Integument. Skin laxity has also been noted in several individuals. Inverted nipples and supernumerary nipples have also been identified in individuals with AKS.
Prognosis. It is unknown whether life span in individuals with AKS is abnormal. One reported individual is alive at age 40 years [Authors, person communication], and the oldest published individual was age 36 years at the time of publication [Choufani et al 2022], demonstrating that survival into adulthood is possible. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with this condition are underrecognized and underreported.
Genotype-Phenotype Correlations
Based on the limited number of reported individuals with AKS, it appears that individuals with missense pathogenic variants may have a reduced burden of major organ malformations compared to individuals with loss-of-function pathogenic variants [Choufani et al 2022]. Additionally, individuals with loss-of-function pathogenic variants (protein truncating or haploinsufficient) are very likely to meet suggested clinical diagnostic criteria, whereas some individuals with missense pathogenic variants may only meet criteria for "possible AKS." This may be due to the fact that all known individuals with loss-of-function pathogenic variants have the recognizable facial gestalt, whereas approximately 70% of individuals with missense pathogenic variants have the recognizable facial gestalt.
General findings to date pertaining to specific features and genotype include the following:
- Microcephaly has only been observed so far in individuals with loss-of-function pathogenic variants.
- Cleft palate. All individuals with cleft palate have had loss-of-function or splice site pathogenic variants; cleft palate has not been reported in individuals with missense pathogenic variants.
- Craniosynostosis. The majority of individuals with craniosynostosis have loss-of-function pathogenic variants, whereas only one individual with a missense pathogenic variant has been reported to have craniosynostosis.
- Congenital heart defects are statistically significantly more common in individuals with loss-of-function pathogenic variants compared to those with missense pathogenic variants (86% vs 23%, respectively).
- High pain tolerance affects about two thirds of individuals with loss-of-function pathogenic variants and one fifth of individuals with missense pathogenic variants.
- Scoliosis is observed in more than two thirds of individuals with loss-of-function pathogenic variants and around 15% of individuals with missense pathogenic variants.
- Vertebral segmentation defects are present in about 10% of individuals with loss-of-function pathogenic variants; these have not been reported in individuals with missense pathogenic variants.
- Hyporeflexia or areflexia is observed in about half of individuals with loss-of-function pathogenic variants and in about 10%-15% of individuals with missense pathogenic variants.
- Developmental delay. The degree of developmental delay may be less severe in individuals with missense pathogenic variants, but evidence for correlating genotype with neurodevelopmental outcomes is still limited.
Nomenclature
The disorder name "HNRNPK-related neurodevelopmental disorder" is based on the dyadic naming approach proposed by Biesecker et al [2021], in which mendelian disorders are designated by combining the mutated gene and resulting phenotype.
Okamoto syndrome was initially described in 1997, although the underlying molecular genetic etiology was unknown [Okamoto et al 1997]. Since then, additional individuals resembling Okamoto syndrome have been identified who share clinical features with AKS and who also have de novo pathogenic variants in HNRNPK. Okamoto syndrome and AKS are therefore now recognized as the same condition [Okamoto 2019, Maystadt et al 2020].
Prevalence
Prevalence is currently unknown. AKS is a rare disorder first described in 2015. The authors are aware of more than 75 affected individuals worldwide, with more than 40 individuals formally reported in the literature [Pua et al 2014, Au et al 2015, Hancarova et al 2015, Lange et al 2016, Miyake et al 2017, Au et al 2018, Dentici et al 2018, Maystadt et al 2020, Choufani et al 2022].
Genetically Related (Allelic) Disorders
Contiguous gene deletions in the 9q21.3 region encompassing HNRNPK and adjacent genes have been reported in individuals who have features of Au-Kline syndrome in addition to other findings attributable to deletion of the adjacent genes [Pua et al 2014, Hancarova et al 2015].
Differential Diagnosis
Table 3.
Disorders to Consider in the Differential Diagnosis of Au-Kline Syndrome
Of note, as individuals with HNRNPK missense pathogenic variants may only have subtle craniofacial differences and relatively few or only minor malformations, mild presentations of Au-Kline syndrome may also now be considered in the differential diagnosis of "nonsyndromic" intellectual disability, particularly in those with hypotonia.
Management
No clinical practice guidelines for Au-Kline syndrome (AKS) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with AKS, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 4.
Au-Kline Syndrome: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
There is no cure for AKS.
Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 5).
Table 5.
Au-Kline Syndrome: Treatment of Manifestations
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Although autism spectrum disorder is uncommon in AKS, children may still qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder (ADHD), when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Prevention of Secondary Complications
Anesthesia consultation is suggested prior to any sedation for surgery given potential airway issues from malocclusion and macroglossia. There is also potential risk that prolonged intubation and ventilation will be required, as occurred in one individual after surgery [Au et al 2018].
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 6 are recommended.
Table 6.
Au-Kline Syndrome: Recommended Surveillance
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Au-Kline syndrome (AKS) is an autosomal dominant disorder typically caused by a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
- All probands reported to date with AKS whose parents have undergone molecular genetic testing have the disorder as the result of a de novo HNRNPK pathogenic variant.
- Molecular genetic testing is recommended for the parents of the proband to evaluate their genetic status and inform recurrence risk assessment.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.* Theoretically, a parent with somatic and germline mosaicism for an HNRNPK pathogenic variant may be mildly/minimally affected.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If the HNRNPK pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk for future sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- Theoretically, if a parent of the proband is known to have the pathogenic variant identified in the proband, the risk to future sibs of inheriting the pathogenic variant is 50%.
Offspring of a proband. To date, individuals with AKS are not known to reproduce; however, many of the reported probands are not yet of reproductive age.
Other family members. Given that all probands with AKS reported to date have the disorder as a result of a de novo HNRNPK pathogenic variant, the risk to other family members is presumed to be low.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of affected individuals.
Prenatal Testing and Preimplantation Genetic Testing
Risk to future pregnancies is presumed to be low as the proband most likely has a de novo HNRNPK pathogenic variant. There is, however, a recurrence risk (~1%) to sibs based on the theoretic possibility of parental germline mosaicism [Rahbari et al 2016]. Given this risk, prenatal and preimplantation genetic testing may be considered.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- HNRNP Family Foundation
- MedlinePlus
- Children's Craniofacial AssociationPhone: 800-535-3643Email: contactCCA@ccakids.com
- FACES: National Craniofacial AssociationPhone: 800-332-2373; 423-266-1632Email: info@faces-cranio.org
- National Institute of Neurological Disorders and Stroke (NINDS)Phone: 800-352-9424
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Au-Kline Syndrome: Genes and Databases
Table B.
OMIM Entries for Au-Kline Syndrome (View All in OMIM)
Molecular Pathogenesis
HNRNPK encodes heterogeneous ribonucleoprotein type K (hnRNP K). There are four isoforms reported in the literature. The longest isoform (isoform a) is 464 amino acids. The alternative protein isoforms have an either basic or acidic C terminus consisting of either 459SGKFF463 or 459ADVEGF464, or they differ due to alternative splicing of exon 8, resulting in missing residues p.Gly111-p.Asn134 [Kimura et al 2010].
HnRNP K is involved with binding single-stranded DNA or RNA, which is mediated through its three KH domains. There are two consecutive KH domains at the N terminus (encoded by exons 4-7 and 9-10) and a third located at the C terminus (encoded by exons 15-16). HnRNP K also acts as a docking platform for interaction of kinases and signal transduction factors that have roles in nucleic acid-related cellular activities, implicating hnRNP K in chromatin remodeling, transcriptional regulation, and translational regulation [Barboro et al 2014].
Mechanism of disease causation. Loss of function
HNRNPK-specific laboratory technical considerations. There are five known cases of AKS with deep intronic pathogenic variants affecting splicing of the gene. Pathogenic variants like this may be missed on exome capture-based platforms.
Cancer and Benign Tumors
Somatic pathogenic variants in HNRNPK have been implicated in human myelodysplasia and leukemia, and mice that are haploinsufficient for HNRNPK are at higher risk for tumors [Barboro et al 2014, Gallardo et al 2015]. However, no hematologic abnormalities or malignancies have been identified in individuals with Au-Kline syndrome. The oldest reported individual is age 40 years. Utility of screening for myelodysplasia is unknown, and therefore no recommendations regarding cancer surveillance can be made.
Chapter Notes
Author Notes
There is an Au-Kline syndrome (AKS) Facebook group for affected individuals and families. Additional support can also be found through the HNRNP Family Foundation.
Dr Ping-Yee Billie Au (ac.sha@ua.eillib) is actively involved in clinical research regarding individuals with AKS. She would be happy to communicate with persons who have any questions regarding diagnosis of AKS or other considerations.
Acknowledgments
We would like to acknowledge the individuals with AKS and their families. We would also like to acknowledge our large group of international collaborators who have contributed to case findings. We would further like to acknowledge the Rare Disease Foundation for providing initial support for phenotype studies. We would like to acknowledge the HNRNP Family Foundation for providing support for patient meetings.
Revision History
- 1 February 2024 (ma) Comprehensive update posted live
- 18 April 2019 (sw) Review posted live
- 15 October 2018 (pyba) Original submission
References
Literature Cited
- Au PYB, Goedhart C, Ferguson M, Breckpot J, Devriendt K, Wierenga K, Fanning E, Grange DK, Graham GE, Galarreta C, Jones MC, Kini U, Stewart H, Parboosingh JS, Kline AD, Innes AM; Care for Rare Canada Consortium. Phenotypic spectrum of Au-Kline syndrome: a report of six new cases and review of the literature. Eur J Hum Genet. 2018;26:1272-81. [PMC free article: PMC6117294] [PubMed: 29904177]
- Au PYB, You J, Caluseriu O, Schwartzentruber J, Majewski J, Bernier FP, Ferguson M; Care for Rare Canada Consortium; Valle D, Parboosingh JS, Sobreira N, Innes AM, Kline AD. GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in HNRNPK. Hum Mutat. 2015;36:1009-14. [PMC free article: PMC4589226] [PubMed: 26173930]
- Barboro P, Ferrari N, Balbi C. Emerging roles of heterogeneous nuclear ribonucleoprotein K (hnRNP K) in cancer progression. Cancer Lett. 2014;352:152-9. [PubMed: 25016060]
- Biesecker LG, Adam MP, Alkuraya FS, Amemiya AR, Bamshad MJ, Beck AE, Bennett JT, Bird LM, Carey JC, Chung B, Clark RD, Cox TC, Curry C, Dinulos MBP, Dobyns WB, Giampietro PF, Girisha KM, Glass IA, Graham JM Jr, Gripp KW, Haldeman-Englert CR, Hall BD, Innes AM, Kalish JM, Keppler-Noreuil KM, Kosaki K, Kozel BA, Mirzaa GM, Mulvihill JJ, Nowaczyk MJM, Pagon RA, Retterer K, Rope AF, Sanchez-Lara PA, Seaver LH, Shieh JT, Slavotinek AM, Sobering AK, Stevens CA, Stevenson DA, Tan TY, Tan WH, Tsai AC, Weaver DD, Williams MS, Zackai E, Zarate YA. A dyadic approach to the delineation of diagnostic entities in clinical genomics. Am J Hum Genet. 2021;108:8-15. [PMC free article: PMC7820621] [PubMed: 33417889]
- Choufani S, McNiven V, Cytrynbaum C, Jangjoo M, Adam MP, Bjornsson HT, Harris J, Dyment DA, Graham GE, Nezarati MM, Aul RB, Castiglioni C, Breckpot J, Devriendt K, Stewart H, Banos-Pinero B, Mehta S, Sandford R, Dunn C, Mathevet R, van Maldergem L, Piard J, Brischoux-Boucher E, Vitobello A, Faivre L, Bournez M, Tran-Mau F, Maystadt I, Fernández-Jaén A, Alvarez S, García-Prieto ID, Alkuraya FS, Alsaif HS, Rahbeeni Z, El-Akouri K, Al-Mureikhi M, Spillmann RC, Shashi V, Sanchez-Lara PA, Graham JM Jr, Roberts A, Chorin O, Evrony GD, Kraatari-Tiri M, Dudding-Byth T, Richardson A, Hunt D, Hamilton L, Dyack S, Mendelsohn BA, Rodríguez N, Sánchez-Martínez R, Tenorio-Castaño J, Nevado J, Lapunzina P, Tirado P, Carminho Amaro Rodrigues MT, Quteineh L, Innes AM, Kline AD, Au PYB, Weksberg R. An HNRNPK-specific DNA methylation signature makes sense of missense variants and expands the phenotypic spectrum of Au-Kline syndrome. Am J Hum Genet. 2022;109:1867-84. [PMC free article: PMC9606382] [PubMed: 36130591]
- Dentici ML, Barresi S, Niceta M, Pantaleoni F, Pizzi S, Dallapiccola B, Tartaglia M, Digilio MC. Clinical spectrum of Kabuki-like syndrome caused by HNRNPK haploinsufficiency. Clin Genet. 2018;93:401-7. [PubMed: 28374925]
- Gallardo M, Lee HJ, Zhang X, Bueso-Ramos C, Pageon LR, McArthur M, Multani A, Nazha A, Manshouri T, Parker-Thornburg J, Rapado I, Quintas-Cardama A, Kornblau SM, Martinez-Lopez J, Post SM. HnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell. 2015;28:486-99. [PMC free article: PMC4652598] [PubMed: 26412324]
- Hancarova M, Puchmajerova A, Drabova J, Karaskova E, Vlckova M, Sedlacek Z. Deletions of 9q21.3 including NTRK2 are associated with severe phenotype. Am J Med Genet A. 2015;167A:264-7. [PubMed: 25348648]
- Kimura Y, Nagata K, Suzuki N, Yokoyama R, Yamanaka Y, Kitamura H, Hirano H, Ohara O. Characterization of multiple alternative forms of heterogeneous nuclear ribonucleoprotein K by phosphate-affinity electrophoresis. Proteomics. 2010;10:3884-95. [PubMed: 20960454]
- Lange L, Pagnamenta AT, Lise S, Clasper S, Stewart H, Akha ES, Quaghebeur G, Knight SJ, Keays DA, Taylor JC, Kini U. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin Genet. 2016;90:258-62. [PMC free article: PMC5006848] [PubMed: 26954065]
- Maystadt I, Deprez M, Moortgat S, Benoît V, Karadurmus D. A second case of Okamoto syndrome caused by HNRNPK mutation. Am J Med Genet A. 2020;182:1537-9. [PubMed: 32222014]
- Miyake N, Inaba M, Mizuno S, Shiina M, Imagawa E, Miyatake S, Nakashima M, Mizuguchi T, Takata A, Ogata K, Matsumoto N. A case of atypical Kabuki syndrome arising from a novel missense variant in HNRNPK. Clin Genet. 2017;92:554-5. [PubMed: 28771707]
- Okamoto N. Okamoto syndrome has features overlapping with Au-Kline syndrome and is caused by HNRNPK mutation. Am J Med Genet A. 2019;179:822-6. [PubMed: 30793470]
- Okamoto N, Matsumoto F, Shimada K, Satomura K. New MCA/MR syndrome with generalized hypotonia, congenital hydronephrosis, and characteristic face. Am J Med Genet. 1997;68:347-9. [PubMed: 9024570]
- Pan X, Liu S, Liu L, Zhang X, Yao H, Tan B. Case report: exome and RNA sequencing identify a novel de novo missense variant in HNRNPK in a Chinese patient with Au-Kline syndrome. Front Genet. 2022;13:853028. [PMC free article: PMC9001983] [PubMed: 35422839]
- Pua HH, Krishnamurthi S, Farrell J, Margeta M, Ursell PC, Powers M, Slavotinek AM, Jeng LJ. Novel interstitial 2.6 Mb deletion on 9q21 associated with multiple congenital anomalies. Am J Med Genet A. 2014;164A:237-42. [PubMed: 24501764]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126-33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
Publication Details
Author Information and Affiliations
Alberta Children's Hospital
University of Calgary
Calgary, Canada
Division of Genetics
McMaster Children's Hospital
Hamilton, Canada
Alberta Children's Hospital
University of Calgary
Calgary, Canada
Alberta Children's Hospital
University of Calgary
Calgary, Canada
Harvey Institute for Human Genetics
Greater Baltimore Medical Center
Towson, Maryland
Publication History
Initial Posting: April 18, 2019; Last Update: February 1, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Au PYB, McNiven V, Phillips L, et al. Au-Kline Syndrome. 2019 Apr 18 [Updated 2024 Feb 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.