Summary
Clinical characteristics.
Saul-Wilson syndrome (SWS) is a skeletal dysplasia characterized by profound short stature, distinctive craniofacial features, short distal phalanges of fingers and toes, and often clubfoot. Early development (primarily speech and motor) is delayed; cognition is normal. Other findings can include hearing loss (conductive, sensorineural, and mixed), lamellar cataracts, and/or rod-cone retinal dystrophy. To date, 16 affected individuals have been reported.
Diagnosis/testing.
The diagnosis of SWS is established in a proband with marked short stature, typical facial and skeletal features, and a heterozygous pathogenic variant in COG4 identified by molecular genetic testing. To date only two COG4 variants, both resulting in a p.Gly516Arg missense change, have been reported.
Management.
Treatment of manifestations: Skeletal dysplasia or physiatry clinic (orthopedist, OT/PT/ rehabilitation specialist) to address repair of clubfoot, possible C1-C2 subluxation and/or spinal cord compression, mobility issues in those with residual foot deformities (post clubfoot repair), osteoarticular pain; standard treatment for feeding issues, speech delay, cataracts and retinal dystrophy, and hearing loss.
Surveillance: Routine follow up of growth and feeding, developmental progress and educational needs, musculoskeletal issues including mobility, osteoarticular pain, bone fragility, possible cataracts and/or retinal dystrophy, hearing loss.
Agents/circumstances to avoid: Participation in gymnastics and jumping on a trampoline until atlanto-axial instability is excluded.
Genetic counseling.
SWS is an autosomal dominant disorder typically caused by a de novo pathogenic variant. Sib recurrence has been observed in one family and is thought to result from germline mosaicism in a parent. The risk to offspring of an individual with SWS of inheriting the COG4 pathogenic variant is 50%. Once the COG4 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
Formal diagnostic criteria for Saul-Wilson syndrome have not been established.
Suggestive Findings
Saul-Wilson syndrome should be suspected in individuals with the following clinical, laboratory, and imaging findings [Ferreira et al 2018].
Clinical findings
- Skeletal
- Profound short stature (typically of prenatal onset)
- Clubfoot
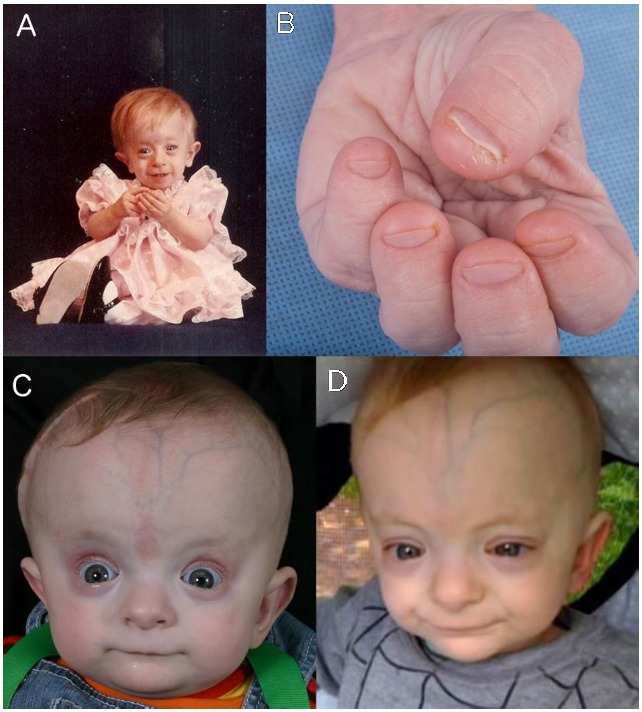
- Short distal phalanges of fingers and toes (See Figure 1.)
- Distinctive craniofacial features (See Figure 1.)
- Progeroid facial appearance (more striking during infancy)
- Sparse hair and sparse eyebrows
- Prominent forehead with prominent scalp veins
- Enlargement and delayed closure of the anterior fontanelle (earliest known closure 21 months; still open at age 3 years in one child)
- Narrow nasal bridge with convex nasal ridge
- Prominent columella (developing in late childhood)
- Thin vermilion of the upper lip
- Mild micrognathia
- Eyes
- Blue sclerae (during the first few months of life)
- Lamellar cataracts
- Rod-cone dystrophy
- Hearing loss (conductive, sensorineural, and mixed)
- Early developmental delay (primarily speech) with normal cognition
Figure 1
A, C, and D. Note prominent forehead, scalp veins, and columella, as well as thin vermilion of the upper lip. B. Note short distal phalanges of the fingers.
Laboratory findings
- Elevated hepatic transaminases
- Intermittent neutropenia
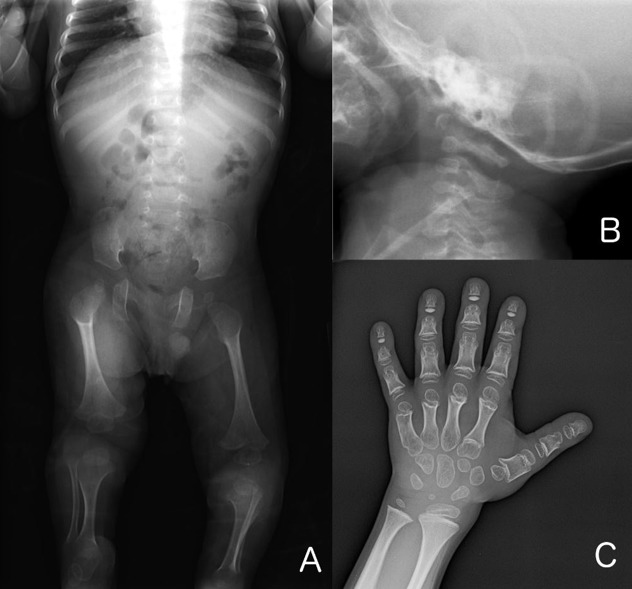
Skeletal radiographs (See Figure 2.)
Figure 2
A. Babygram obtained at age 11 months. Note coxa valga, overtubulation of the long bones, and lucencies of the proximal femora. B. Lateral cervical spine radiograph obtained at age eight months highlighting hypoplasia of the odontoid process
- Long bones
- Short long bones
- Overtubulation with thin diaphyses and flared metaphyses
- Lucency of proximal femora
- Coxa valga
- Megaepiphyses
- Hand
- Small hands with short metacarpals and short phalanges
- Accessory ossification centers of the proximal metacarpals
- Cone-shaped epiphyses of the phalanges
- Ivory epiphyses of the distal phalanges (in late childhood)
- Spine
- Hypoplasia of the odontoid process of C2
- Hypoplasia of T12 or L1
- Platyspondyly (vertebral bodies become taller with age)
- Irregularities of the endplates of the vertebral bodies (in later life)
Establishing the Diagnosis
The diagnosis of Saul-Wilson syndrome is established in a proband with marked short stature, typical facial and skeletal features, and a heterozygous pathogenic (or likely pathogenic) variant in COG4 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a heterozygous COG4 variant of uncertain significance does not establish or rule out the diagnosis.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because Saul-Wilson syndrome is rare, individuals with the distinctive findings described in Suggestive Findings in whom the diagnosis is recognized are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of Saul-Wilson syndrome has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1: Single-Gene Testing
Sequence analysis of COG4 detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
Note: (1) Since only two variants, both resulting in a p.Gly516Arg missense change, have been reported to date, targeted analysis for these variants could be performed first to confirm a clinical diagnosis of Saul-Wilson syndrome. (2) Since Saul-Wilson syndrome occurs through a gain-of-function mechanism and large intragenic deletion or duplication has not been reported, testing for intragenic deletions or duplication is unlikely to identify a disease-causing variant.
Option 2: Genomic Testing
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Saul-Wilson Syndrome
Clinical Characteristics
Clinical Description
Saul-Wilson syndrome is a skeletal dysplasia characterized by profound short stature, distinctive craniofacial features, short distal phalanges of fingers and toes, and often clubfoot. Early development (primarily speech) is delayed; cognition is normal. Other findings can include hearing loss (conductive, sensorineural, and mixed), lamellar cataracts, and/or rod-cone retinal dystrophy.
A total of 16 individuals with Saul-Wilson syndrome have been reported to date. Saul-Wilson syndrome was first described in a small-for-gestational-age infant with bulging fontanelles, clubfoot, blue sclerae, and blunted fingertips; over time, growth was delayed and the child developed bilateral cataracts, and hearing loss as the result of frequent otitis media [Saul 1982]. Three additional individuals with similar features were reported [Saul & Wilson 1990, Hersh et al 1994]. Subsequently the diagnosis of Saul-Wilson syndrome was entertained in a child without typical facial features [Chinen et al 2015], but the diagnosis could not be confirmed by molecular genetic testing. Fourteen individuals were described in 2018, including two originally reported in the 1990s [Ferreira et al 2018]. Since that publication, a few additional individuals with Saul-Wilson syndrome worldwide have been diagnosed [Author, personal observation]. The clinical findings discussed in this section are based on these reports.
Growth
Individuals with Saul-Wilson syndrome show impaired postnatal growth, and several also had intrauterine growth restriction (IUGR).
Mean length, weight, and head circumference at birth:
- Length. 44.1 cm (range: 38.0-49.0)
- Weight. 2.09 kg (range: 1.45-2.80)
- Head circumference. 31.7 cm (range: 29.0-34.0)
Z scores at birth:
- Length. -2.3 (1.5 SD; range: -0.4 to -5.1)
- Weight. -2.4 (0.7 SD; range: -1.2 to -3.8)
- Head circumference: -2.0 (0.9 SD; range: -0.8 to -3.9)
Z scores decline sharply over the first few months of life. At last examination:
- Stature. -6.3 (1.8 SD; range: -3.5 to -9.8)
- Weight. -4.0 (1.2 SD; range: -1.1 to -5.8)
- Head circumference. -1.7 (1.7 SD; range: 0.8 to -5.0)
Based on data available from three adults
- Mean height, weight, and head circumference at skeletal maturity:
- Height. 107.6 cm
- Weight. 30.5 kg
- Head circumference. 50.2 cm
- Z scores at skeletal maturity:
- Height. -8.9 (0.8 SD; range: -8.3 to -9.8)
- Weight. -4.3 (0.6 SD; range: -3.6 to -4.8)
- Head circumference. -3.9 (1.6 SD; range: -2.7 to -5.0)
Growth charts for clinical use are currently under development.
Despite absolute microcephaly, head circumferences exceed the height by more than 2 SD, with consequent relative macrocephaly.
Development
Speech delay (8/11) and motor delay (12/14) are common, probably related to the presence of hearing loss and skeletal deformities, respectively; cognitive development does not appear to be affected.
Ophthalmologic Features
The majority of affected individuals develop lamellar cataracts during the first few years of life (10/13), and several developed retinal involvement (6/9). Retinal pigmentary changes can be seen in the periphery as early as the toddler years. During adolescence and early adulthood, a rod-cone dystrophy (5/9) becomes evident with constricted visual fields and night blindness.
Macular cystic changes were also described (2/4). One individual had myelinated retinal nerve fibers [Ferreira et al 2020].
Skeletal Features
Shortening of the distal phalanges of fingers and toes is appreciated on physical examination (12/14). This finding, apparent at birth, did not progress over time. The majority of individuals (10/14) had clubfoot, and in some cases residual deformity even after multiple attempts at surgical repair [Ferreira et al 2020]. Pectus deformity (5/14) and cervical spinal cord compression (3/7) have also been reported.
Bone fragility has been suggested, as several individuals (4/14) developed fractures with minimal or no known trauma [Ferreira et al 2020]. One of these individuals had poor fracture healing, which was also described in the original patient [Saul & Wilson 1990]. Nonunion with pseudoarthrosis has been seen in two individuals [Saul & Wilson 1990, Ferreira et al 2020]. Although DXA scans for two individuals reported bone mineral density (BMD) <2 SD below the mean, the height-adjusted BMD [Zemel et al 2010, Zemel et al 2011] was normal in both [Author, personal observation].
Osteoarticular pain was reported by all three adults. Two had confirmed degenerative joint disease, leading to joint replacement surgeries in one individual in her 20s [Ferreira et al 2020].
The combination of megaepiphyses with coxa valga, leading to acetabulum-femoral epiphyseal incongruence, may contribute to premature osteoarthropathy of the hip.
Other
Hearing loss, seen in the majority of affected individuals over time, can be conductive, sensorineural, or mixed. In one child hearing impairment associated with inner-ear malformations was detected on newborn hearing screen.
MRI findings. Ventriculomegaly was seen in 5/9 and spinal cord syrinx in 1/4 individuals. Spinal cord compression was observed in 3/7: in one child with soft-tissue pannus surrounding the odontoid process, it was seen as early as age four years, and in another child with cervical spine instability, as late as age 14 years.
Intermittent neutropenia, though not appreciated in the first two months of life, was seen in all 12 individuals subsequently evaluated for this finding [Ferreira et al 2020]. The earliest known age of onset is three months, and it still occurred in adults. While intermittent neutropenia could be one possible explanation for the frequent (although rarely life-threatening) respiratory infections experienced in the first years of life, the neutropenia persisted into adulthood whereas the number of respiratory infections decreased over time.
Asymptomatic elevation of liver transaminases. Elevated aspartate aminotransferase was observed in 6/8 individuals and elevated alanine aminotransferase in 3/8, without abnormalities in other liver function tests, such as serum albumin, coagulation parameters, alkaline phosphatase, and bilirubin [Ferreira et al 2020].
Genotype-Phenotype Correlations
No genotype-phenotype correlation is possible because all affected individuals have the same amino acid substitution.
Penetrance
Penetrance is thought to be 100%.
Prevalence
Sixteen individuals have been reported to date. Three additional individuals are known to have the diagnosis of Saul-Wilson syndrome [Author, personal observation]. There is no known geographic predilection.
Genetically Related (Allelic) Disorders
Biallelic loss-of-function or hypomorphic variants in COG4 are associated with COG4-CDG (see Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview). COG4-CDG manifests with seizures, hypotonia, intellectual disability, microcephaly, elevated transaminases, and in one case recurrent infections.
Differential Diagnosis
Table 2.
Disorders Interest in the Differential Diagnosis of Saul-Wilson Syndrome
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Saul-Wilson syndrome, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Saul-Wilson Syndrome
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with Saul-Wilson Syndrome
Developmental Delay Management Issues
The following information represents typical management recommendations for individuals with developmental delay in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Surveillance
Table 5.
Recommended Surveillance for Individuals with Saul-Wilson Syndrome
Agents/Circumstances to Avoid
Participation in gymnastics and jumping on a trampoline should be avoided until atlanto-axial instability is excluded.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnancy of an affected woman has not been documented to date, although regular menstrual cycles and a normal hormone profile in two adult females suggest no impairment of the ability to conceive [Ferreira et al 2020].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Saul-Wilson syndrome is an autosomal dominant disorder typically caused by a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
- No individual with Saul-Wilson syndrome reported to date has had an affected parent.
- Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant.
- If the COG4 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the pathogenic variant most likely occurred de novo in the proband. Another possible explanation is germline mosaicism in a parent. Parental germline mosaicism is presumed in one family with two affected sibs.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the COG4 pathogenic variant identified in the proband, the risk to the sibs is 50%.
- If the proband has a known SWS-related COG4 pathogenic variant that cannot be detected in the leukocyte DNA of either parent or the parents have not been tested for the COG4 pathogenic variant but are clinically unaffected, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with SWS has a 50% chance of inheriting the SWS-related COG4 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the pathogenic variant identified in the proband or clinical evidence of the disorder, the pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
Prenatal Testing and Preimplantation Genetic Testing
Once the COG4 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- National Organization for Rare Disorders (NORD)Phone: 800-999-6673
- Skeletal Dysplasia Management Consortium
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Saul-Wilson Syndrome: Genes and Databases
Table B.
OMIM Entries for Saul-Wilson Syndrome (View All in OMIM)
Molecular Pathogenesis
COG4 is a subunit of the conserved oligomeric Golgi (COG) complex, a hetero-octameric protein complex that regulates vesicular trafficking between the Golgi apparatus and the endoplasmic reticulum (ER).
Mechanism of disease causation. The distinct phenotypes, recurrent nature of the p.Gly516Arg variant, and accelerated (not delayed) retrograde Golgi-to-ER transport seen in cells support Saul-Wilson syndrome occurring through a gain-of-function mechanism [Ferreira et al 2018].
Table 6.
Notable COG4 Pathogenic Variants
Chapter Notes
Revision History
- 20 February 2020 (bp) Review posted live
- 4 October 2019 (cf) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "Saul-Wilson Syndrome" is in the public domain in the United States of America.
References
Literature Cited
- Chinen Y, Kaneshi T, Kamiya T, Hata K, Nishimura G, Kaname T. Progressive hip joint subluxation in Saul-Wilson syndrome. Am J Med Genet A. 2015;167A:2834–8. [PubMed: 26239279]
- Ferreira CR, Xia ZJ, Clément A, Parry DA, Davids M, Taylan F, Sharma P, Turgeon CT, Blanco-Sánchez B, Ng BG, Logan CV, Wolfe LA, Solomon BD, Cho MT, Douglas G, Carvalho DR, Bratke H, Haug MG, Phillips JB, Wegner J, Tiemeyer M, Aoki K, Nordgren A, Hammarsjö A, Duker AL, Rohena L, Hove HB, Ek J, Adams D, Tifft CJ, Onyekweli T, Weixel T, Macnamara E, Radtke K, Powis Z, Earl D, Gabriel M, Russi AHS, Brick L, Kozenko M, Tham E, Raymond KM, Phillips JA 3rd, Tiller GE, Wilson WG, Hamid R, Malicdan MCV, Nishimura G, Grigelioniene G, Jackson A, Westerfield M, Bober MB, Gahl WA, Freeze HH, et al. A recurrent de novo heterozygous COG4 substitution leads to Saul-Wilson syndrome, disrupted vesicular trafficking, and altered proteoglycan glycosylation. Am J Hum Genet. 2018;103:553–67. [PMC free article: PMC6174323] [PubMed: 30290151]
- Ferreira CR, Zein WM, Huryn LA, Merker A, Berger SI, Wilson WG, Tiller GE, Wolfe LA, Merideth M, Carvalho DR, Duker AL, Bratke H, Haug MG, Rohena L, Hove HB, Xia ZJ, Ng BG, Freeze HH, Gabriel M, Russi AHS, Brick L, Kozenko M, Earl DL, Tham E, Nishimura G, Phillips JA 3rd, Gahl WA, Hamid R, Jackson AP, Grigelioniene G, Bober MB. Defining the clinical phenotype of Saul-Wilson syndrome. Genet Med. 2020;22:857–66. [PMC free article: PMC7205587] [PubMed: 31949312]
- Hersh JH, Joyce MR, Spranger J, Goatley EC, Lachman RS, Bhatt S, Rimoin DL. Microcephalic osteodysplastic dysplasia. Am J Med Genet. 1994;51:194–9. [PubMed: 8074143]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Saul RA. Unknown cases. Proc Greenwood Genet Ctr. 1982;1:102–5.
- Saul RA, Wilson WG. A new skeletal dysplasia in two unrelated boys. Am J Med Genet. 1990;35:388–93. [PubMed: 2309787]
- White KK, Bompadre V, Goldberg MJ, Bober MB, Cho TJ, Hoover-Fong JE, Irving M, Mackenzie WG, Kamps SE, Raggio C, Redding GJ, Spencer SS, Savarirayan R, Theroux MC, et al. Best practices in peri-operative management of patients with skeletal dysplasias. Am J Med Genet A. 2017;173:2584–95. [PubMed: 28763154]
- Zemel BS, Kalkwarf HJ, Gilsanz V, Lappe JM, Oberfield S, Shepherd JA, Frederick MM, Huang X, Lu M, Mahboubi S, Hangartner T, Winer KK. Revised reference curves for bone mineral content and areal bone mineral density according to age and sex for black and non-black children: results of the bone mineral density in childhood study. J Clin Endocrinol Metab. 2011;96:3160–9. [PMC free article: PMC3200252] [PubMed: 21917867]
- Zemel BS, Leonard MB, Kelly A, Lappe JM, Gilsanz V, Oberfield S, Mahboubi S, Shepherd JA, Hangartner TN, Frederick MM, Winer KK, Kalkwarf HJ. Height adjustment in assessing dual energy x-ray absorptiometry measurements of bone mass and density in children. J Clin Endocrinol Metab. 2010;95:1265–73. [PMC free article: PMC2841534] [PubMed: 20103654]
Publication Details
Author Information and Affiliations
National Human Genome Research Institute
National Institutes of Health
Bethesda, Maryland
Publication History
Initial Posting: February 20, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Ferreira C. Saul-Wilson Syndrome. 2020 Feb 20. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.